

ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2
- PMID: 20460269
- PMCID: PMC2901139
- DOI: 10.1093/hmg/ddq202
ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2
Abstract
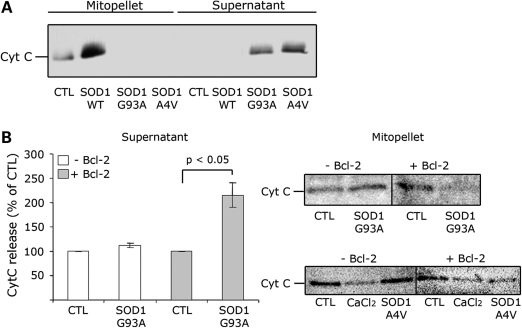
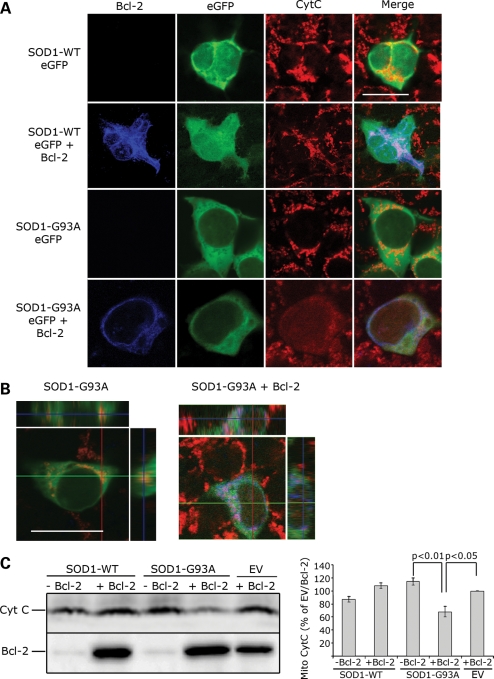
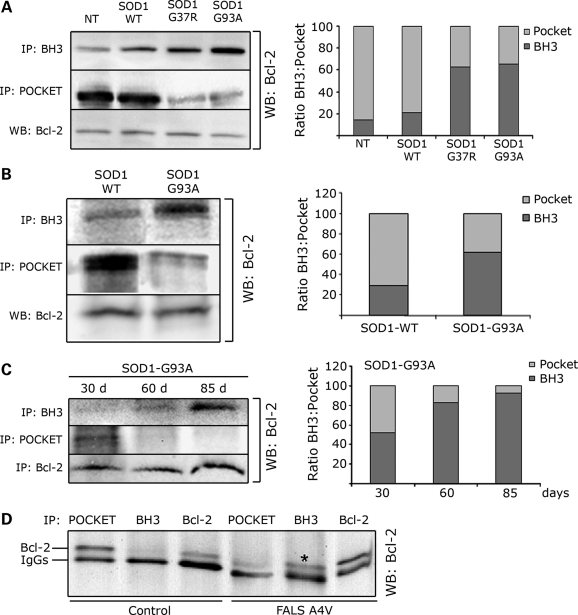
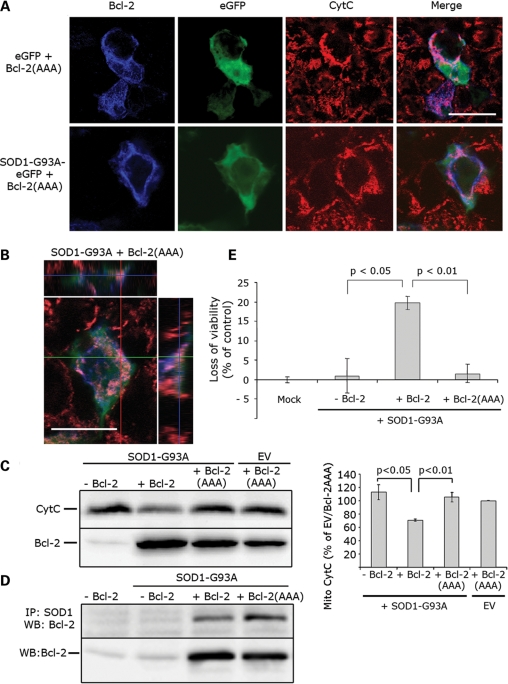
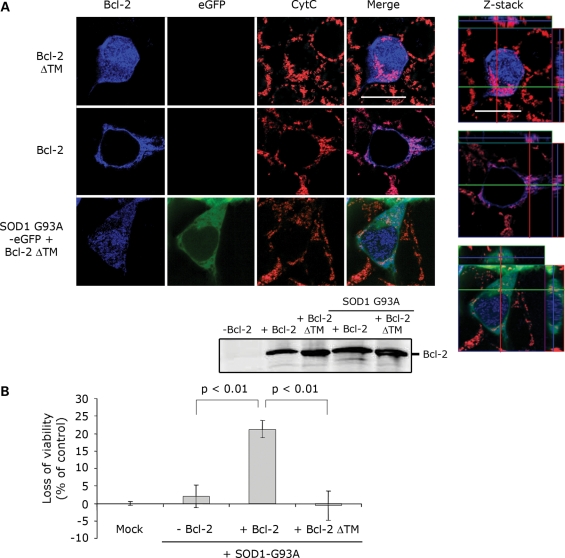
In mutant superoxide dismutase (SOD1)-linked amyotrophic lateral sclerosis (ALS), accumulation of misfolded mutant SOD1 in spinal cord mitochondria is thought to cause mitochondrial dysfunction. Whether mutant SOD1 is toxic per se or whether it damages the mitochondria through interactions with other mitochondrial proteins is not known. We previously identified Bcl-2 as an interacting partner of mutant SOD1 specifically in spinal cord, but not in liver, mitochondria of SOD1 mice and patients. We now show that mutant SOD1 toxicity relies on this interaction. Mutant SOD1 induces mitochondrial morphological changes and compromises mitochondrial membrane integrity leading to release of Cytochrome C only in the presence of Bcl-2. In cells, mouse and human spinal cord with SOD1 mutations, the binding to mutant SOD1 triggers a conformational change in Bcl-2 that results in the uncovering of its toxic BH3 domain and conversion of Bcl-2 into a toxic protein. Bcl-2 carrying a mutagenized, non-toxic BH3 domain fails to support mutant SOD1 mitochondrial toxicity. The identification of Bcl-2 as a specific target and active partner in mutant SOD1 mitochondrial toxicity suggests new therapeutic strategies to inhibit the formation of the toxic mutant SOD1/Bcl-2 complex and to prevent mitochondrial damage in ALS.
Figures

Similar articles
-
Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis.Biochim Biophys Acta. 2014 Aug;1842(8):1295-301. doi: 10.1016/j.bbadis.2014.02.009. Epub 2014 Feb 22. Biochim Biophys Acta. 2014. PMID: 24568860 Free PMC article. Review.
-
The Ile35 Residue of the ALS-Associated Mutant SOD1 Plays a Crucial Role in the Intracellular Aggregation of the Molecule.Mol Neurobiol. 2025 Feb;62(2):2023-2038. doi: 10.1007/s12035-024-04369-0. Epub 2024 Jul 26. Mol Neurobiol. 2025. PMID: 39060907
-
SOD1 exhibits allosteric frustration to facilitate metal binding affinity.Proc Natl Acad Sci U S A. 2013 Mar 5;110(10):3871-6. doi: 10.1073/pnas.1216597110. Epub 2013 Feb 19. Proc Natl Acad Sci U S A. 2013. PMID: 23431152 Free PMC article.
-
Identification of a novel toxicophore in anti-cancer chemotherapeutics that targets mitochondrial respiratory complex I.Elife. 2020 May 20;9:e55845. doi: 10.7554/eLife.55845. Elife. 2020. PMID: 32432547 Free PMC article.
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
Cited by
-
The Role of Bioenergetics in Neurodegeneration.Int J Mol Sci. 2022 Aug 16;23(16):9212. doi: 10.3390/ijms23169212. Int J Mol Sci. 2022. PMID: 36012480 Free PMC article. Review.
-
Sex specific activation of the ERα axis of the mitochondrial UPR (UPRmt) in the G93A-SOD1 mouse model of familial ALS.Hum Mol Genet. 2017 Apr 1;26(7):1318-1327. doi: 10.1093/hmg/ddx049. Hum Mol Genet. 2017. PMID: 28186560 Free PMC article.
-
Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons.J Neurosci. 2012 Jan 4;32(1):229-42. doi: 10.1523/JNEUROSCI.1233-11.2012. J Neurosci. 2012. PMID: 22219285 Free PMC article.
-
The 2-Oxoglutarate Carrier Is S-Nitrosylated in the Spinal Cord of G93A Mutant hSOD1 Mice Resulting in Disruption of Mitochondrial Glutathione Transport.Biomedicines. 2022 Dec 27;11(1):61. doi: 10.3390/biomedicines11010061. Biomedicines. 2022. PMID: 36672568 Free PMC article.
-
In vivo and in vitro determination of cell death markers in neurons.Methods Mol Biol. 2011;793:9-21. doi: 10.1007/978-1-61779-328-8_2. Methods Mol Biol. 2011. PMID: 21913091 Free PMC article.
References
-
- Pasinelli P., Brown R.H. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat. Rev. Neurosci. 2006;7:710–723. doi:10.1038/nrn1971. - DOI - PubMed
-
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi:10.1038/362059a0. - DOI - PubMed
-
- Jaarsma D., Rognoni F., van Duijn W., Verspaget H.W., Haasdijk E.D., Holstege J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. - PubMed
-
- Liu J., Lillo C., Jonsson P.A., Vande Velde C., Ward C.M., Miller T.M., Subramaniam J.R., Rothstein J.D., Marklund S., Andersen P.M., et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi:10.1016/j.neuron.2004.06.016. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
