

Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer's disease
- PMID: 20413848
- PMCID: PMC4996661
- DOI: 10.3233/JAD-2010-100306
Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer's disease
Abstract
Alzheimer's disease (AD) is the most common neurodegenerative disorder among the aged worldwide. AD is characterized by extensive synaptic and neuronal loss that leads to impaired memory and cognitive decline. The cause of AD is not completely understood and no effective therapy has been developed. The accumulation of toxic amyloid-beta42 (Abeta42) peptide oligomers and aggregates in AD brain has been proposed to be primarily responsible for the pathology of the disease, an idea dubbed the 'amyloid hypothesis' of AD etiology. In addition to the increase in Abeta42 levels, disturbances in neuronal calcium (Ca2+) signaling and alterations in expression levels of Ca2+ signaling proteins have been observed in animal models of familial AD and in studies of postmortem brain samples from sporadic AD patients. Based on these data, the 'Ca2+ hypothesis of AD' has been proposed. In particular, familial AD has been linked with enhanced Ca2+ release from the endoplasmic reticulum and elevated cytosolic Ca2+ levels. The augmented cytosolic Ca2+ levels can trigger signaling cascades that affect synaptic stability and function and can be detrimental to neuronal health, such as activation of calcineurin and calpains. Here we review the latest results supporting the 'Ca2+ hypothesis' of AD pathogenesis. We further argue that over time, supranormal cytosolic Ca2+ signaling can impair mitochondrial function in AD neurons. We conclude that inhibitors and stabilizers of neuronal Ca2+ signaling and mitochondrial function may have therapeutic potential for AD treatment. We also discuss latest and planned AD therapeutic trials of agents targeting Ca2+ channels and mitochondria.
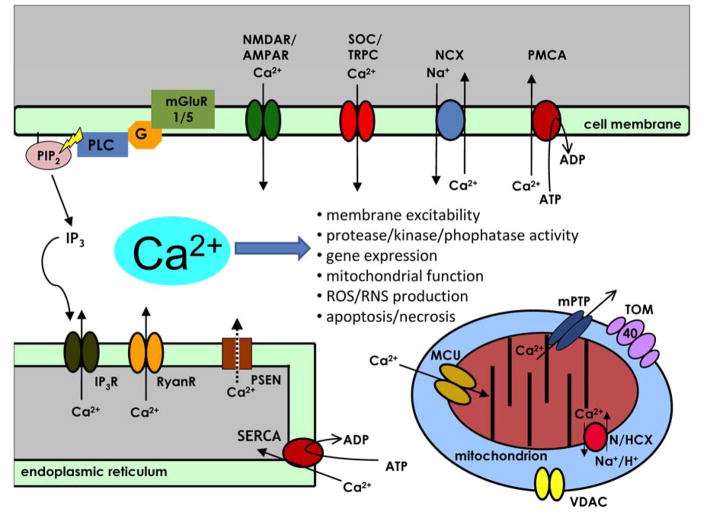
Figures
Similar articles
-
The dysregulation of intracellular calcium in Alzheimer disease.Cell Calcium. 2010 Feb;47(2):183-9. doi: 10.1016/j.ceca.2009.12.014. Epub 2010 Jan 18. Cell Calcium. 2010. PMID: 20080301 Free PMC article. Review.
-
Presenilins as endoplasmic reticulum calcium leak channels and Alzheimer's disease pathogenesis.Sci China Life Sci. 2011 Aug;54(8):744-51. doi: 10.1007/s11427-011-4201-y. Epub 2011 Jul 24. Sci China Life Sci. 2011. PMID: 21786197 Review.
-
Mitochondrial Calcium Signaling as a Therapeutic Target for Alzheimer's Disease.Curr Alzheimer Res. 2020;17(4):329-343. doi: 10.2174/1567205016666191210091302. Curr Alzheimer Res. 2020. PMID: 31820698 Review.
-
Presenilins function in ER calcium leak and Alzheimer's disease pathogenesis.Cell Calcium. 2011 Sep;50(3):303-9. doi: 10.1016/j.ceca.2011.05.013. Epub 2011 Jun 12. Cell Calcium. 2011. PMID: 21663966 Free PMC article. Review.
-
Reversal of Calcium Dysregulation as Potential Approach for Treating Alzheimer's Disease.Curr Alzheimer Res. 2020;17(4):344-354. doi: 10.2174/1567205017666200528162046. Curr Alzheimer Res. 2020. PMID: 32469698 Free PMC article. Review.
Cited by
-
Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction.Front Aging Neurosci. 2015 Jun 10;7:101. doi: 10.3389/fnagi.2015.00101. eCollection 2015. Front Aging Neurosci. 2015. PMID: 26113818 Free PMC article. Review.
-
Relationships between Mitochondrial Dysfunction and Neurotransmission Failure in Alzheimer's Disease.Aging Dis. 2020 Oct 1;11(5):1291-1316. doi: 10.14336/AD.2019.1125. eCollection 2020 Oct. Aging Dis. 2020. PMID: 33014538 Free PMC article. Review.
-
Investigating Pathogenetic Mechanisms of Alzheimer's Disease by Systems Biology Approaches for Drug Discovery.Int J Mol Sci. 2021 Oct 19;22(20):11280. doi: 10.3390/ijms222011280. Int J Mol Sci. 2021. PMID: 34681938 Free PMC article.
-
Evaluation of the adaptogenic potential exerted by ginsenosides Rb1 and Rg1 against oxidative stress-mediated neurotoxicity in an in vitro neuronal model.PLoS One. 2017 Aug 16;12(8):e0182933. doi: 10.1371/journal.pone.0182933. eCollection 2017. PLoS One. 2017. PMID: 28813475 Free PMC article.
-
The role of Bcl-2 proteins in modulating neuronal Ca2+ signaling in health and in Alzheimer's disease.Biochim Biophys Acta Mol Cell Res. 2021 May;1868(6):118997. doi: 10.1016/j.bbamcr.2021.118997. Epub 2021 Mar 9. Biochim Biophys Acta Mol Cell Res. 2021. PMID: 33711363 Free PMC article. Review.
References
-
- Bachurin S, Bukatina E, Lermontova N, Tkachenko S, Afanasiev A, Grigoriev V, Grigorieva I, Ivanov Y, Sablin S, Zefirov N. Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer. Annals of the New York Academy of Sciences. 2001;939:425–435. - PubMed
-
- Bachurin SO, Shevtsova EP, Kireeva EG, Oxenkrug GF, Sablin SO. Mitochondria as a target for neurotoxins and neuroprotective agents. Annals of the New York Academy of Sciences. 2003;993:334–344. discussion 345-339. - PubMed
-
- Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis. 2006;9:119–126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
