

Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability
- PMID: 20392936
- PMCID: PMC3073615
- DOI: 10.1523/JNEUROSCI.5711-09.2010
Cholecystokinin facilitates glutamate release by increasing the number of readily releasable vesicles and releasing probability
Abstract
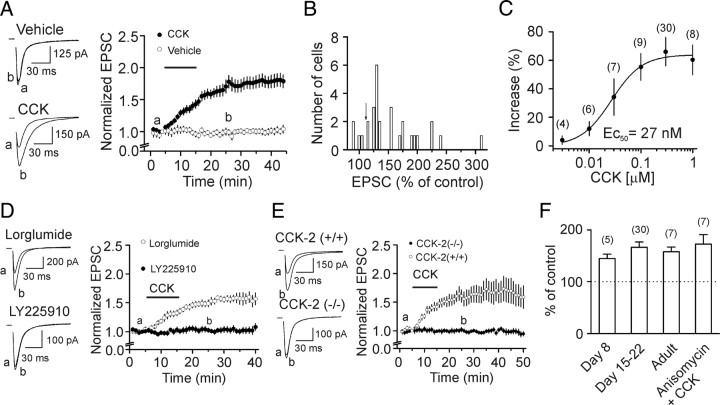
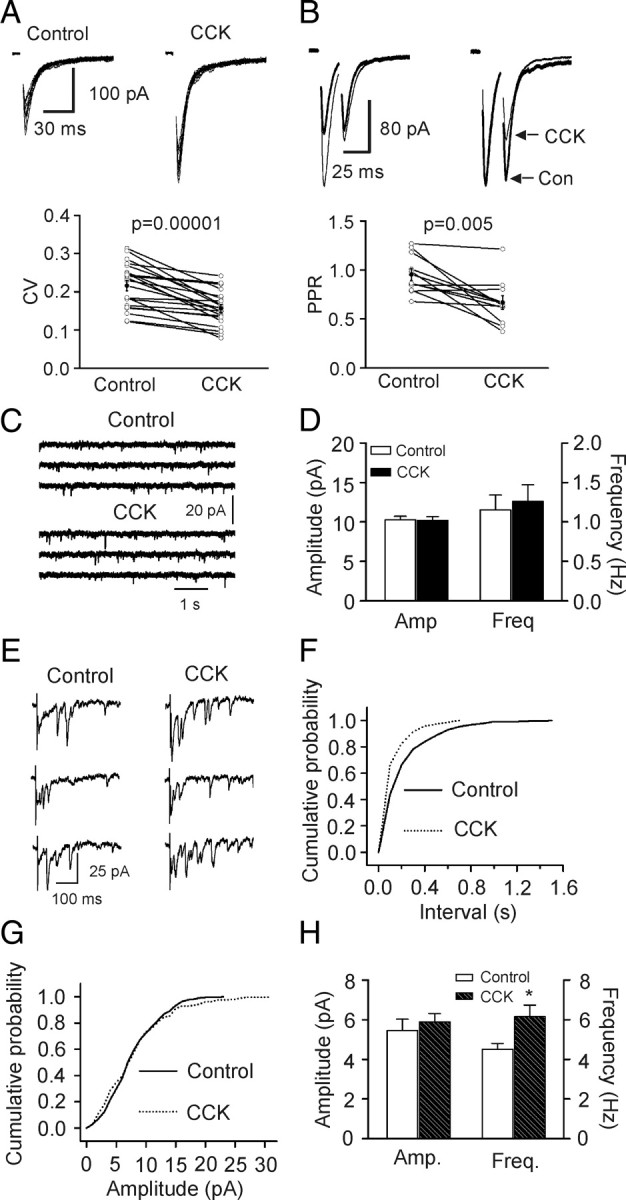
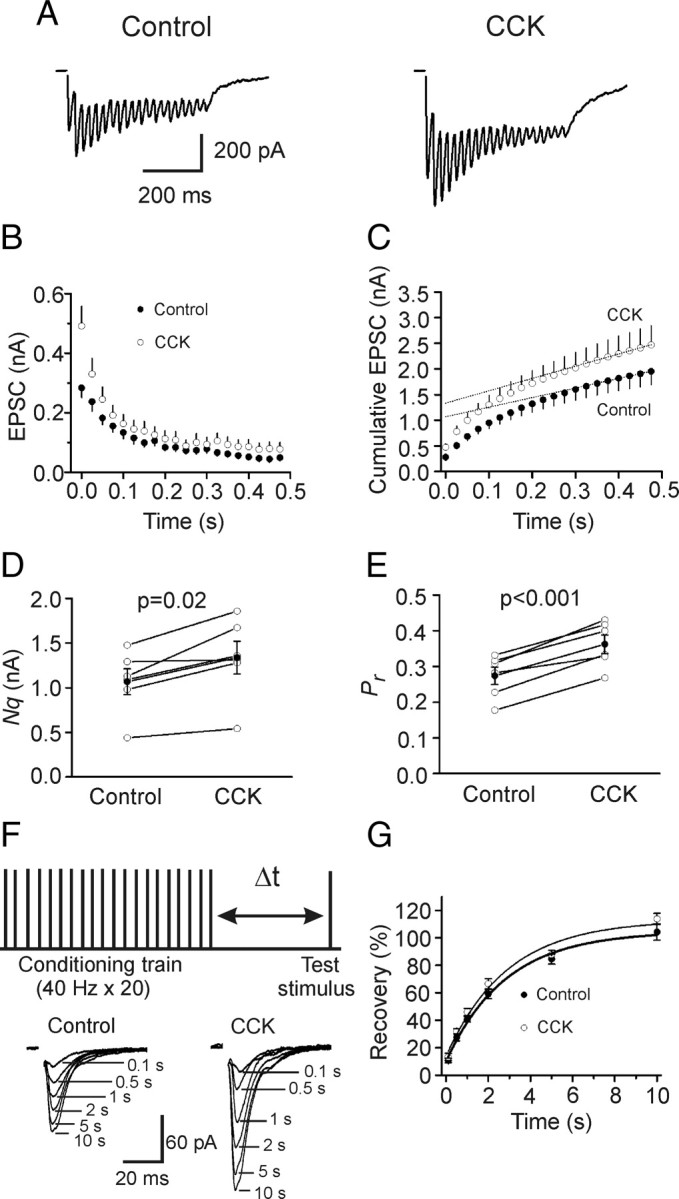
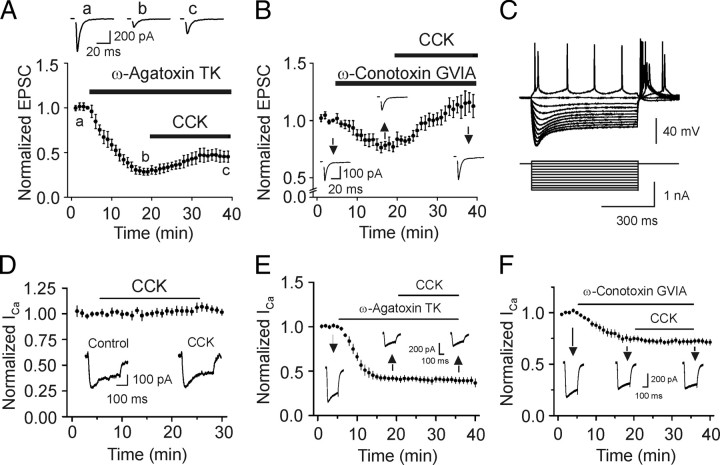
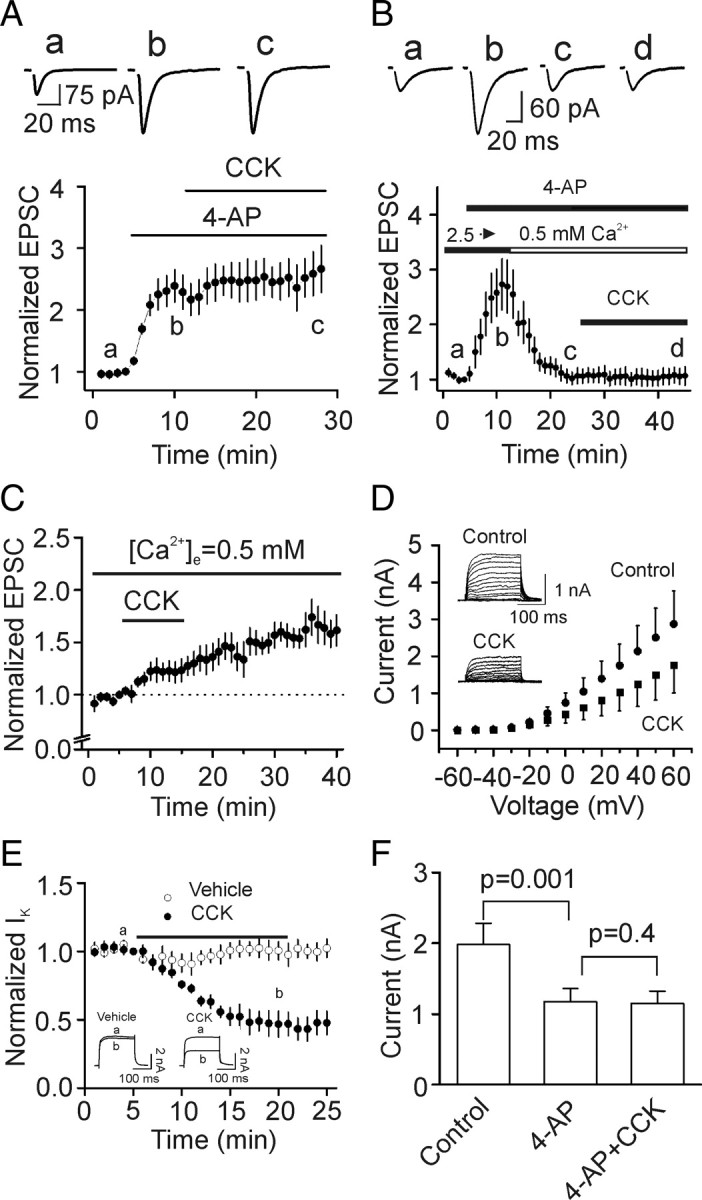
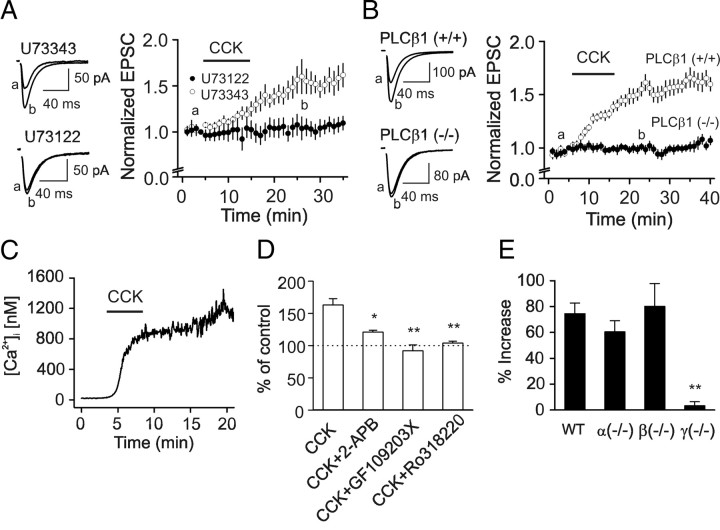
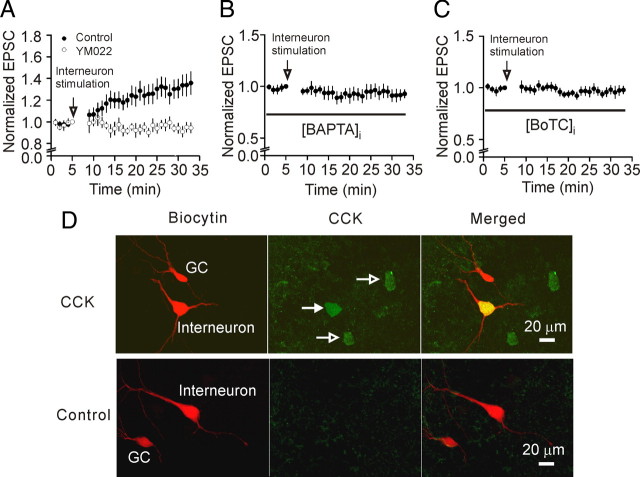
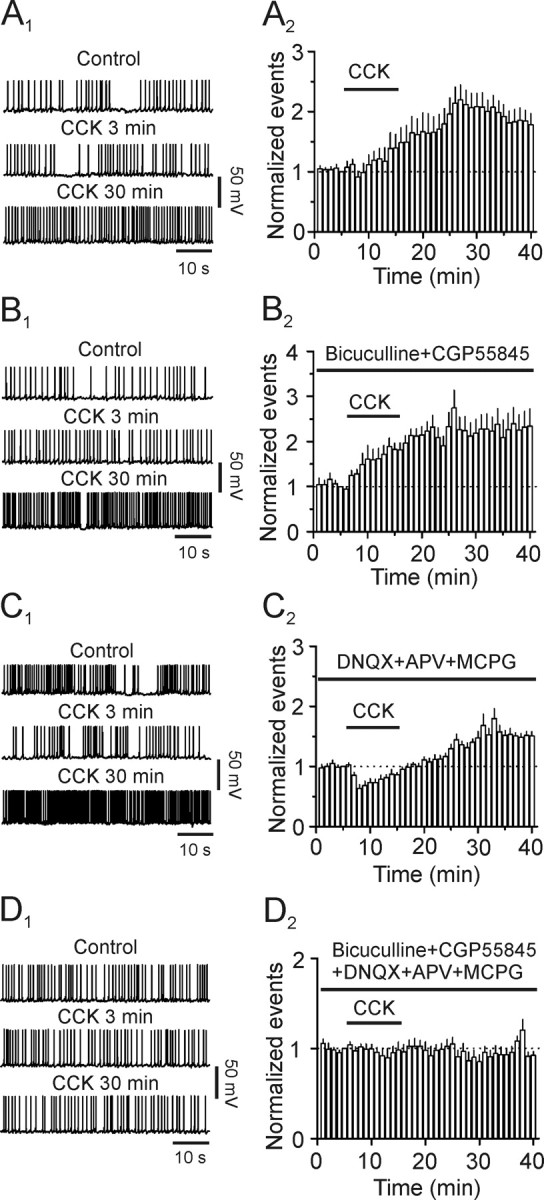
Cholecystokinin (CCK), a neuropeptide originally discovered in the gastrointestinal tract, is abundantly distributed in the mammalian brains including the hippocampus. Whereas CCK has been shown to increase glutamate concentration in the perfusate of hippocampal slices and in purified rat hippocampal synaptosomes, the cellular and molecular mechanisms whereby CCK modulates glutamatergic function remain unexplored. Here, we examined the effects of CCK on glutamatergic transmission in the hippocampus using whole-cell recordings from hippocampal slices. Application of CCK increased AMPA receptor-mediated EPSCs at perforant path-dentate gyrus granule cell, CA3-CA3 and Schaffer collateral-CA1 synapses without effects at mossy fiber-CA3 synapses. CCK-induced increases in AMPA EPSCs were mediated by CCK-2 receptors and were not modulated developmentally and transcriptionally. CCK reduced the coefficient of variation and paired-pulse ratio of AMPA EPSCs suggesting that CCK facilitates presynaptic glutamate release. CCK increased the release probability and the number of readily releasable vesicles with no effects on the rate of recovery from vesicle depletion. CCK-mediated increases in glutamate release required the functions of phospholipase C, intracellular Ca(2+) release and protein kinase Cgamma. CCK released endogenously from hippocampal interneurons facilitated glutamatergic transmission. Our results provide a cellular and molecular mechanism to explain the roles of CCK in the brain.
Figures
Similar articles
-
Neurotensinergic augmentation of glutamate release at the perforant path-granule cell synapse in rat dentate gyrus: Roles of L-Type Ca²⁺ channels, calmodulin and myosin light-chain kinase.Neuropharmacology. 2015 Aug;95:252-60. doi: 10.1016/j.neuropharm.2015.03.028. Epub 2015 Apr 2. Neuropharmacology. 2015. PMID: 25842242 Free PMC article.
-
Bidirectional modulation of GABAergic transmission by cholecystokinin in hippocampal dentate gyrus granule cells of juvenile rats.J Physiol. 2006 Apr 15;572(Pt 2):425-42. doi: 10.1113/jphysiol.2005.104463. Epub 2006 Feb 2. J Physiol. 2006. PMID: 16455686 Free PMC article.
-
GABA B receptor modulation of excitatory and inhibitory synaptic transmission onto rat CA3 hippocampal interneurons.J Physiol. 2003 Jan 15;546(Pt 2):439-53. doi: 10.1113/jphysiol.2002.034017. J Physiol. 2003. PMID: 12527730 Free PMC article.
-
Role of giant depolarizing potentials in shaping synaptic currents in the developing hippocampus.Crit Rev Neurobiol. 2006;18(1-2):13-23. doi: 10.1615/critrevneurobiol.v18.i1-2.30. Crit Rev Neurobiol. 2006. PMID: 17725505 Review.
-
Shaping excitation at glutamatergic synapses.Trends Neurosci. 1999 Oct;22(10):451-8. doi: 10.1016/s0166-2236(99)01445-9. Trends Neurosci. 1999. PMID: 10481192 Review.
Cited by
-
Vasopressin facilitates GABAergic transmission in rat hippocampus via activation of V(1A) receptors.Neuropharmacology. 2012 Dec;63(7):1218-26. doi: 10.1016/j.neuropharm.2012.07.043. Epub 2012 Aug 3. Neuropharmacology. 2012. PMID: 22884625 Free PMC article.
-
Involvement of TRPC5 channels, inwardly rectifying K+ channels, PLCβ and PIP2 in vasopressin-mediated excitation of medial central amygdala neurons.J Physiol. 2021 Jun;599(12):3101-3119. doi: 10.1113/JP281260. Epub 2021 Apr 27. J Physiol. 2021. PMID: 33871877 Free PMC article.
-
Phospholipase C not protein kinase C is required for the activation of TRPC5 channels by cholecystokinin.Eur J Pharmacol. 2012 Aug 15;689(1-3):17-24. doi: 10.1016/j.ejphar.2012.05.032. Epub 2012 Jun 7. Eur J Pharmacol. 2012. PMID: 22683873 Free PMC article.
-
Requirement of phospholipase C and protein kinase C in cholecystokinin-mediated facilitation of NMDA channel function and anxiety-like behavior.Hippocampus. 2012 Jun;22(6):1438-50. doi: 10.1002/hipo.20984. Epub 2011 Nov 10. Hippocampus. 2012. PMID: 22072552 Free PMC article.
-
Upregulation of transmitter release probability improves a conversion of synaptic analogue signals into neuronal digital spikes.Mol Brain. 2012 Aug 1;5:26. doi: 10.1186/1756-6606-5-26. Mol Brain. 2012. PMID: 22852823 Free PMC article.
References
-
- Barrie AP, Nicholls DG, Sanchez-Prieto J, Sihra TS. An ion channel locus for the protein kinase C potentiation of transmitter glutamate release from guinea pig cerebrocortical synaptosomes. J Neurochem. 1991;57:1398–1404. - PubMed
-
- Beinfeld MC. An introduction to neuronal cholecystokinin. Peptides. 2001;22:1197–1200. - PubMed
-
- Bergink V, van Megen HJ, Westenberg HG. Glutamate and anxiety. Eur Neuropsychopharmacol. 2004;14:175–183. - PubMed
-
- Boden PR, Hill RG. Effects of cholecystokinin and pentagastrin on rat hippocampal neurones maintained in vitro. Neuropeptides. 1988;12:95–103. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous