

Super-resolution biomolecular crystallography with low-resolution data
- PMID: 20376006
- PMCID: PMC2859093
- DOI: 10.1038/nature08892
Super-resolution biomolecular crystallography with low-resolution data
Abstract
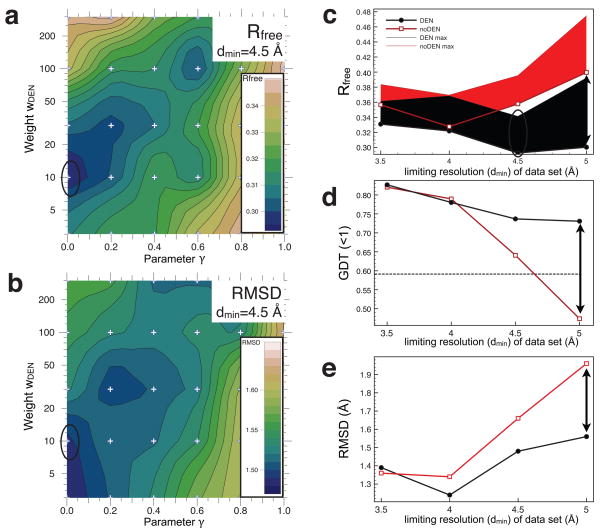
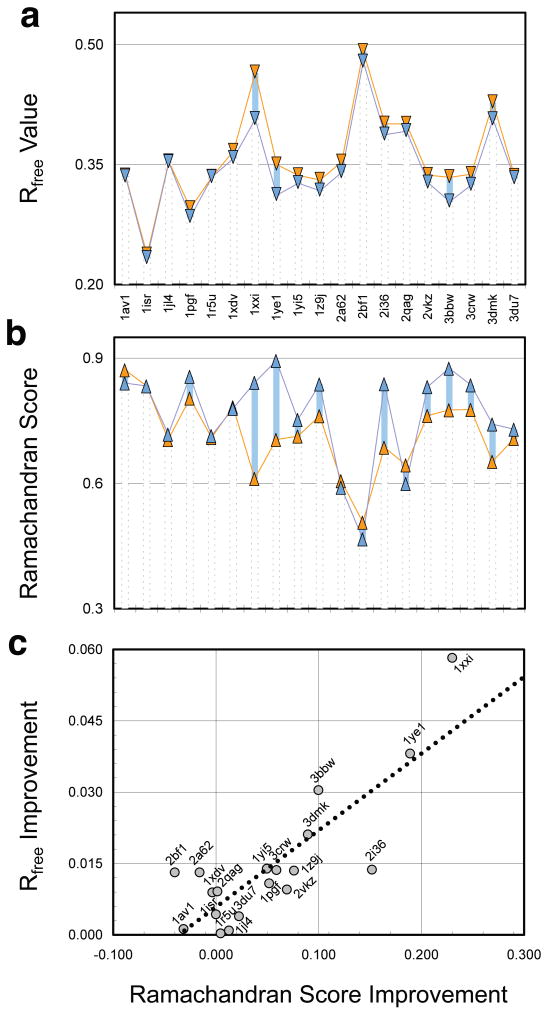
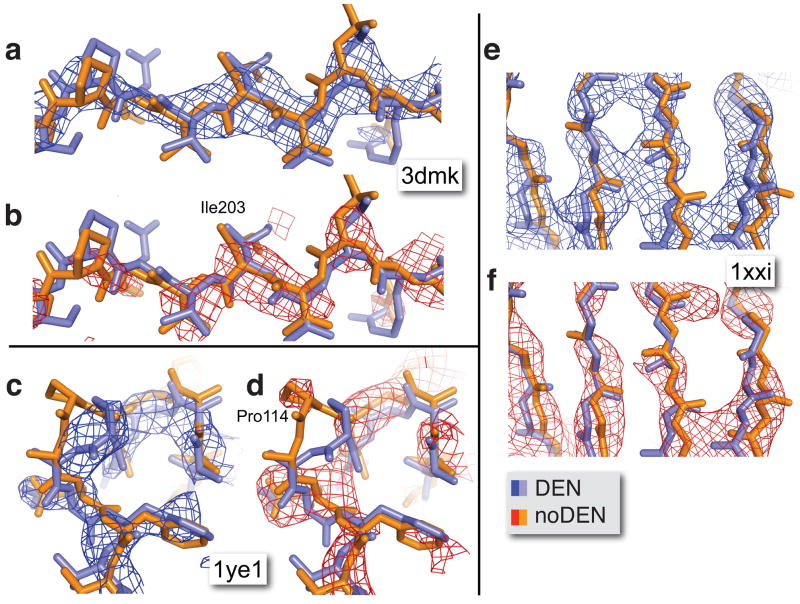
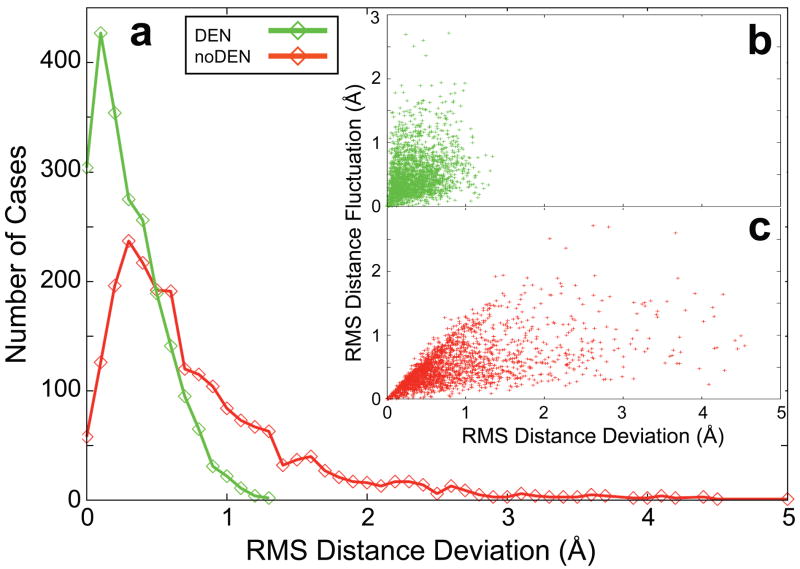
X-ray diffraction plays a pivotal role in the understanding of biological systems by revealing atomic structures of proteins, nucleic acids and their complexes, with much recent interest in very large assemblies like the ribosome. As crystals of such large assemblies often diffract weakly (resolution worse than 4 A), we need methods that work at such low resolution. In macromolecular assemblies, some of the components may be known at high resolution, whereas others are unknown: current refinement methods fail as they require a high-resolution starting structure for the entire complex. Determining the structure of such complexes, which are often of key biological importance, should be possible in principle as the number of independent diffraction intensities at a resolution better than 5 A generally exceeds the number of degrees of freedom. Here we introduce a method that adds specific information from known homologous structures but allows global and local deformations of these homology models. Our approach uses the observation that local protein structure tends to be conserved as sequence and function evolve. Cross-validation with R(free) (the free R-factor) determines the optimum deformation and influence of the homology model. For test cases at 3.5-5 A resolution with known structures at high resolution, our method gives significant improvements over conventional refinement in the model as monitored by coordinate accuracy, the definition of secondary structure and the quality of electron density maps. For re-refinements of a representative set of 19 low-resolution crystal structures from the Protein Data Bank, we find similar improvements. Thus, a structure derived from low-resolution diffraction data can have quality similar to a high-resolution structure. Our method is applicable to the study of weakly diffracting crystals using X-ray micro-diffraction as well as data from new X-ray light sources. Use of homology information is not restricted to X-ray crystallography and cryo-electron microscopy: as optical imaging advances to subnanometre resolution, it can use similar tools.
Conflict of interest statement
AUTHOR INFORMATION
The authors declare no competing financial interests.
Figures
Comment in
-
From poor resolution to rich insight.Structure. 2010 Jun 9;18(6):664-5. doi: 10.1016/j.str.2010.05.004. Structure. 2010. PMID: 20541503
Similar articles
-
Strategies for crystallization and structure determination of very large macromolecular assemblies.Curr Opin Struct Biol. 2007 Oct;17(5):572-9. doi: 10.1016/j.sbi.2007.09.004. Epub 2007 Oct 25. Curr Opin Struct Biol. 2007. PMID: 17964135 Review.
-
Low Resolution Refinement of Atomic Models Against Crystallographic Data.Methods Mol Biol. 2017;1607:565-593. doi: 10.1007/978-1-4939-7000-1_23. Methods Mol Biol. 2017. PMID: 28573589 Review.
-
Determining the Crystal Structure of TRPV6.In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. In: Kozak JA, Putney JW Jr, editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL): CRC Press/Taylor & Francis; 2018. Chapter 14. PMID: 30299652 Free Books & Documents. Review.
-
A Practical Approach to Protein Crystallography.Methods Mol Biol. 2017;1525:47-78. doi: 10.1007/978-1-4939-6622-6_3. Methods Mol Biol. 2017. PMID: 27896717
-
Transferability of multipole charge-density parameters: application to very high resolution oligopeptide and protein structures.Acta Crystallogr D Biol Crystallogr. 1998 Nov 1;54(Pt 6 Pt 2):1306-18. doi: 10.1107/s0907444998004466. Acta Crystallogr D Biol Crystallogr. 1998. PMID: 10089507
Cited by
-
Bacterial chemoreceptor arrays are hexagonally packed trimers of receptor dimers networked by rings of kinase and coupling proteins.Proc Natl Acad Sci U S A. 2012 Mar 6;109(10):3766-71. doi: 10.1073/pnas.1115719109. Epub 2012 Feb 21. Proc Natl Acad Sci U S A. 2012. PMID: 22355139 Free PMC article.
-
PIM: phase integrated method for normal mode analysis of biomolecules in a crystalline environment.J Mol Biol. 2013 Mar 25;425(6):1082-98. doi: 10.1016/j.jmb.2012.12.026. Epub 2013 Jan 16. J Mol Biol. 2013. PMID: 23333742 Free PMC article.
-
A novel N-acetylglutamate synthase architecture revealed by the crystal structure of the bifunctional enzyme from Maricaulis maris.PLoS One. 2011;6(12):e28825. doi: 10.1371/journal.pone.0028825. Epub 2011 Dec 12. PLoS One. 2011. PMID: 22174908 Free PMC article.
-
Noncanonical protein kinase A activation by oligomerization of regulatory subunits as revealed by inherited Carney complex mutations.Proc Natl Acad Sci U S A. 2021 May 25;118(21):e2024716118. doi: 10.1073/pnas.2024716118. Proc Natl Acad Sci U S A. 2021. PMID: 34006641 Free PMC article.
-
SAXS ensemble refinement of ESCRT-III CHMP3 conformational transitions.Structure. 2011 Jan 12;19(1):109-16. doi: 10.1016/j.str.2010.10.006. Structure. 2011. PMID: 21220121 Free PMC article.
References
-
- Davies JM, Brunger AT, Weis WI. Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure. 2008;16 (5):715–726. - PubMed
-
- Raines KS, et al. Three-dimensional structure determination from a single view. Nature. 2010;463:214–217. - PubMed
-
- Pertsinidis A, Zhang Y, Chu S. Localization, registration and distance measurements between single-molecule fluorescent probes with sub-nanometer precision and accuracy. Nature. 2010 sutmitted. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
