

RNA-mediated neurodegeneration in repeat expansion disorders
- PMID: 20373340
- PMCID: PMC2852186
- DOI: 10.1002/ana.21948
RNA-mediated neurodegeneration in repeat expansion disorders
Abstract
Most neurodegenerative disorders are thought to result primarily from the accumulation of misfolded proteins, which interfere with protein homeostasis in neurons. For a subset of diseases, however, noncoding regions of RNAs assume a primary toxic gain-of-function, leading to degeneration in many tissues, including the nervous system. Here we review a series of proposed mechanisms by which noncoding repeat expansions give rise to nervous system degeneration and dysfunction. These mechanisms include transcriptional alterations and the generation of antisense transcripts, sequestration of mRNA-associated protein complexes that lead to aberrant mRNA splicing and processing, and alterations in cellular processes, including activation of abnormal signaling cascades and failure of protein quality control pathways. We place these potential mechanisms in the context of known RNA-mediated disorders, including the myotonic dystrophies and fragile X tremor ataxia syndrome, and discuss recent results suggesting that mRNA toxicity may also play a role in some presumably protein-mediated neurodegenerative disorders. Lastly, we comment on recent progress in therapeutic development for these RNA-dominant diseases.
Figures
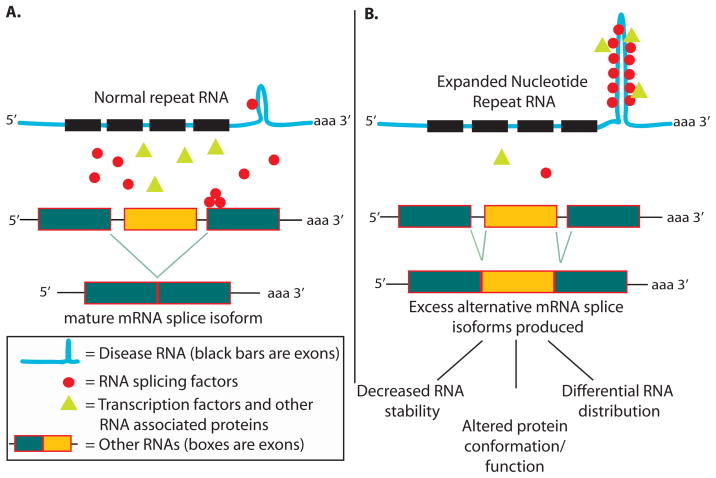
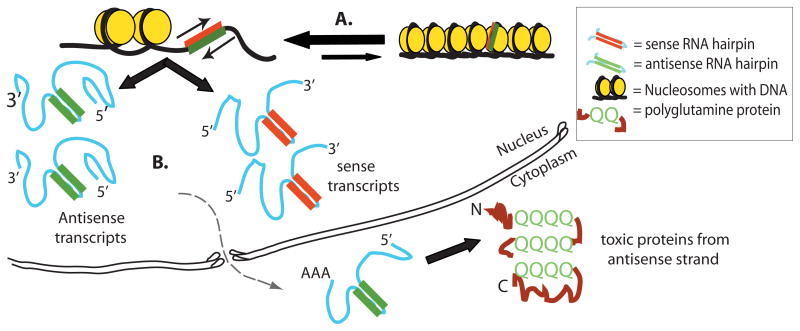
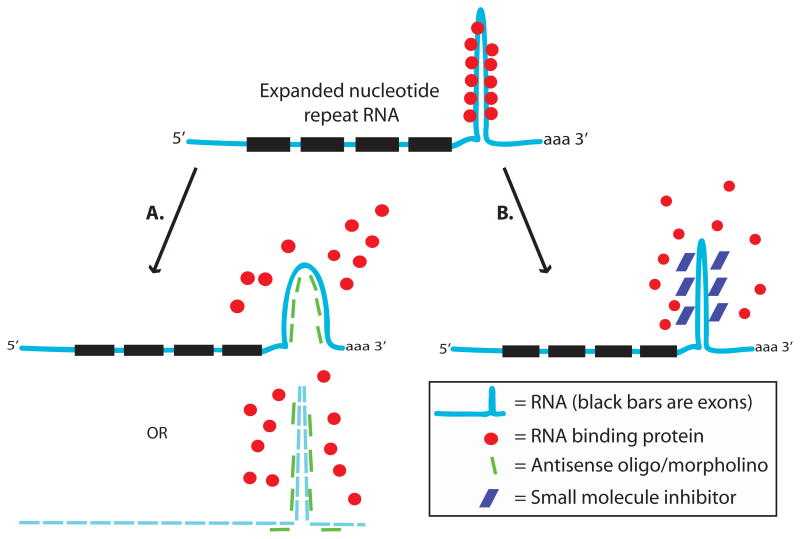
Similar articles
-
CAG repeat RNA as an auxiliary toxic agent in polyglutamine disorders.RNA Biol. 2011 Jul-Aug;8(4):565-71. doi: 10.4161/rna.8.4.15397. Epub 2011 Jul 1. RNA Biol. 2011. PMID: 21593608 Free PMC article. Review.
-
Repeat RNA expansion disorders of the nervous system: post-transcriptional mechanisms and therapeutic strategies.Crit Rev Biochem Mol Biol. 2021 Feb;56(1):31-53. doi: 10.1080/10409238.2020.1841726. Epub 2020 Nov 10. Crit Rev Biochem Mol Biol. 2021. PMID: 33172304 Free PMC article. Review.
-
Trinucleotide repeat disorders.Annu Rev Neurosci. 2007;30:575-621. doi: 10.1146/annurev.neuro.29.051605.113042. Annu Rev Neurosci. 2007. PMID: 17417937 Review.
-
Gain of RNA function in pathological cases: Focus on myotonic dystrophy.Biochimie. 2011 Nov;93(11):2006-12. doi: 10.1016/j.biochi.2011.06.028. Epub 2011 Jul 13. Biochimie. 2011. PMID: 21763392 Review.
-
RNA-dominant diseases.Hum Mol Genet. 2006 Oct 15;15 Spec No 2:R162-9. doi: 10.1093/hmg/ddl181. Hum Mol Genet. 2006. PMID: 16987879 Review.
Cited by
-
Non-canonical DNA/RNA structures associated with the pathogenesis of Fragile X-associated tremor/ataxia syndrome and Fragile X syndrome.Front Genet. 2022 Aug 30;13:866021. doi: 10.3389/fgene.2022.866021. eCollection 2022. Front Genet. 2022. PMID: 36110216 Free PMC article. Review.
-
Mutant huntingtin messenger RNA forms neuronal nuclear clusters in rodent and human brains.Brain Commun. 2022 Oct 13;4(6):fcac248. doi: 10.1093/braincomms/fcac248. eCollection 2022. Brain Commun. 2022. PMID: 36458209 Free PMC article.
-
Oligonucleotide-Based Therapy for FTD/ALS Caused by the C9orf72 Repeat Expansion: A Perspective.J Nucleic Acids. 2013;2013:208245. doi: 10.1155/2013/208245. Epub 2013 Nov 17. J Nucleic Acids. 2013. PMID: 24349764 Free PMC article. Review.
-
RNA phase transitions in repeat expansion disorders.Nature. 2017 Jun 8;546(7657):243-247. doi: 10.1038/nature22386. Epub 2017 May 31. Nature. 2017. PMID: 28562589 Free PMC article.
-
SFPQ regulates the accumulation of RNA foci and dipeptide repeat proteins from the expanded repeat mutation in C9orf72.J Cell Sci. 2021 Feb 19;134(4):jcs256602. doi: 10.1242/jcs.256602. J Cell Sci. 2021. PMID: 33495278 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
