

Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes
- PMID: 20351235
- PMCID: PMC2946079
- DOI: 10.1161/CIRCULATIONAHA.109.914911
Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes
Abstract
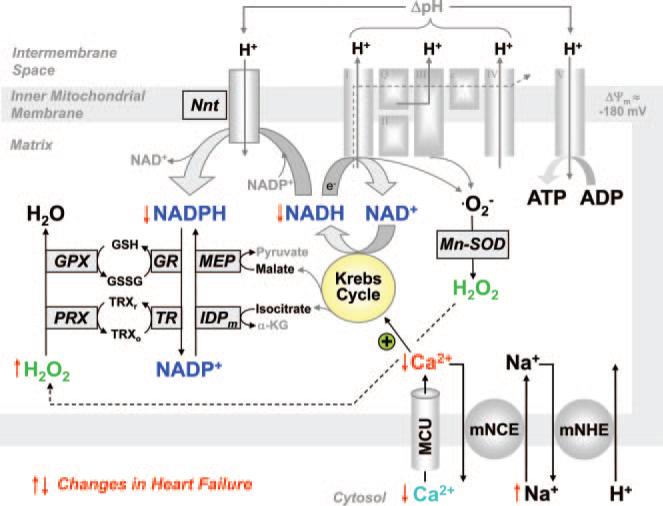
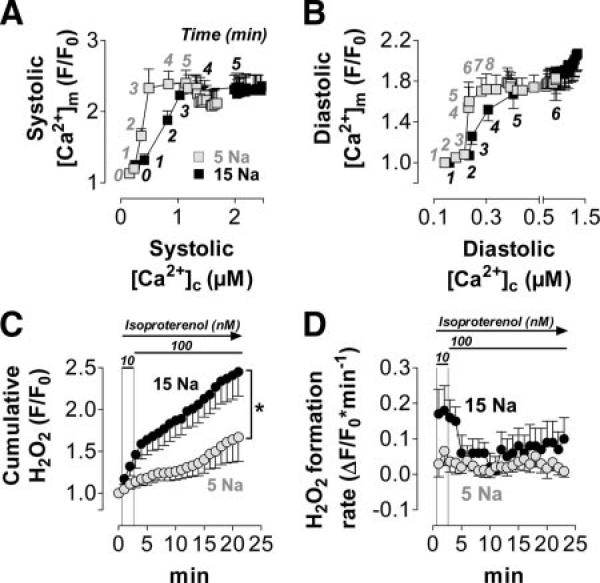
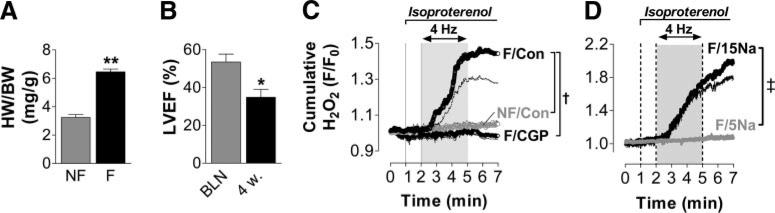
Background: Oxidative stress is causally linked to the progression of heart failure, and mitochondria are critical sources of reactive oxygen species in failing myocardium. We previously observed that in heart failure, elevated cytosolic Na(+) ([Na(+)](i)) reduces mitochondrial Ca(2+) ([Ca(2+)](m)) by accelerating Ca(2+) efflux via the mitochondrial Na(+)/Ca(2+) exchanger. Because the regeneration of antioxidative enzymes requires NADPH, which is indirectly regenerated by the Krebs cycle, and Krebs cycle dehydrogenases are activated by [Ca(2+)](m), we speculated that in failing myocytes, elevated [Na(+)](i) promotes oxidative stress.
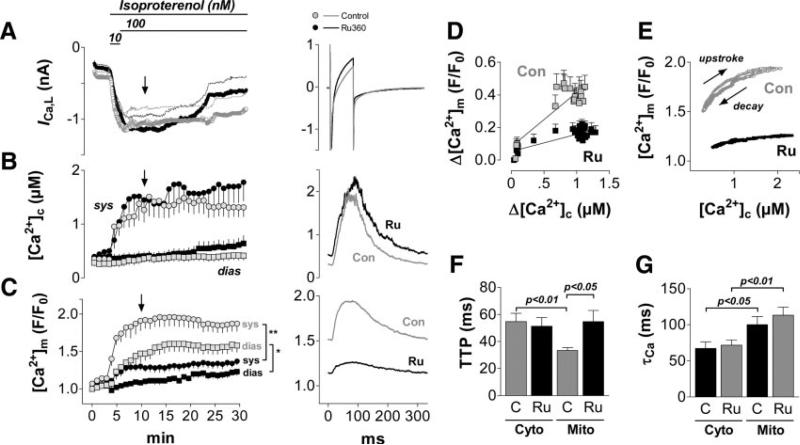
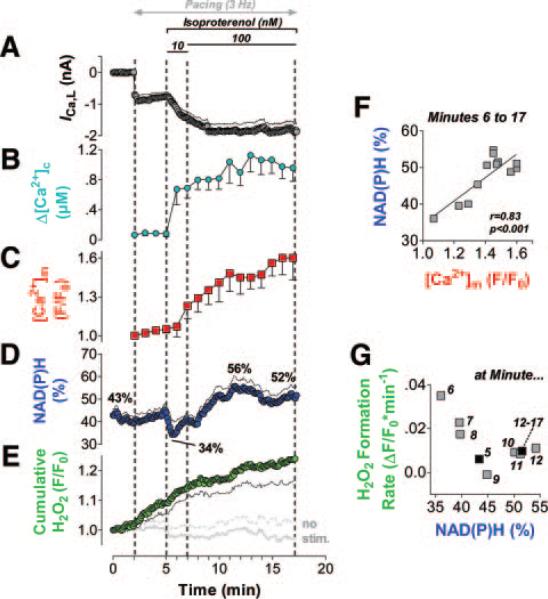
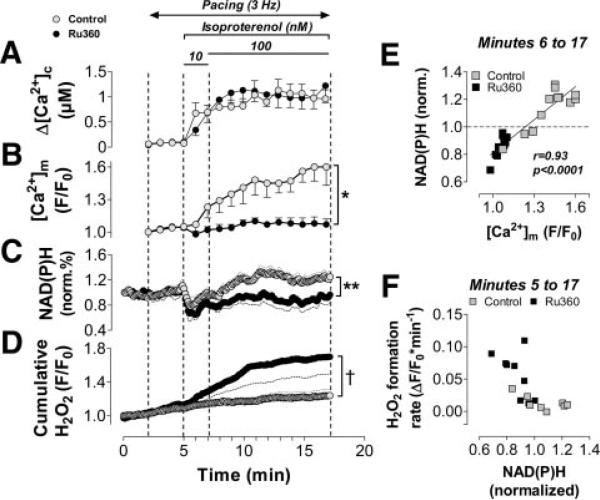
Methods and results: We used a patch-clamp-based approach to simultaneously monitor cytosolic and mitochondrial Ca(2+) and, alternatively, mitochondrial H(2)O(2) together with NAD(P)H in guinea pig cardiac myocytes. Cells were depolarized in a voltage-clamp mode (3 Hz), and a transition of workload was induced by beta-adrenergic stimulation. During this transition, NAD(P)H initially oxidized but recovered when [Ca(2+)](m) increased. The transient oxidation of NAD(P)H was closely associated with an increase in mitochondrial H(2)O(2) formation. This reactive oxygen species formation was potentiated when mitochondrial Ca(2+) uptake was blocked (by Ru360) or Ca(2+) efflux was accelerated (by elevation of [Na(+)](i)). In failing myocytes, H(2)O(2) formation was increased, which was prevented by reducing mitochondrial Ca(2+) efflux via the mitochondrial Na(+)/Ca(2+) exchanger.
Conclusions: Besides matching energy supply and demand, mitochondrial Ca(2+) uptake critically regulates mitochondrial reactive oxygen species production. In heart failure, elevated [Na(+)](i) promotes reactive oxygen species formation by reducing mitochondrial Ca(2+) uptake. This novel mechanism, by which defects in ion homeostasis induce oxidative stress, represents a potential drug target to reduce reactive oxygen species production in the failing heart.
Figures
Similar articles
-
Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes.Circ Res. 2006 Jul 21;99(2):172-82. doi: 10.1161/01.RES.0000232546.92777.05. Epub 2006 Jun 15. Circ Res. 2006. PMID: 16778127 Free PMC article.
-
Adverse bioenergetic consequences of Na+-Ca2+ exchanger-mediated Ca2+ influx in cardiac myocytes.Circulation. 2010 Nov 30;122(22):2273-80. doi: 10.1161/CIRCULATIONAHA.110.968057. Epub 2010 Nov 15. Circulation. 2010. PMID: 21098439
-
Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching.Circ Res. 2008 Aug 1;103(3):279-88. doi: 10.1161/CIRCRESAHA.108.175919. Epub 2008 Jul 3. Circ Res. 2008. PMID: 18599868 Free PMC article.
-
Calcium Signaling and Reactive Oxygen Species in Mitochondria.Circ Res. 2018 May 11;122(10):1460-1478. doi: 10.1161/CIRCRESAHA.118.310082. Circ Res. 2018. PMID: 29748369 Review.
-
Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes.Free Radic Biol Med. 2013 Oct;63:338-49. doi: 10.1016/j.freeradbiomed.2013.05.035. Epub 2013 Jun 1. Free Radic Biol Med. 2013. PMID: 23732518 Review.
Cited by
-
Benefits of SGLT2 inhibitors in arrhythmias.Front Cardiovasc Med. 2022 Oct 20;9:1011429. doi: 10.3389/fcvm.2022.1011429. eCollection 2022. Front Cardiovasc Med. 2022. PMID: 36337862 Free PMC article. Review.
-
Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities.Free Radic Biol Med. 2018 Dec;129:155-168. doi: 10.1016/j.freeradbiomed.2018.09.019. Epub 2018 Sep 15. Free Radic Biol Med. 2018. PMID: 30227272 Free PMC article. Review.
-
An integrated mitochondrial ROS production and scavenging model: implications for heart failure.Biophys J. 2013 Dec 17;105(12):2832-42. doi: 10.1016/j.bpj.2013.11.007. Biophys J. 2013. PMID: 24359755 Free PMC article.
-
The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca2+ transient amplitude in rabbit failing myocytes.Pflugers Arch. 2018 Jun;470(6):959-968. doi: 10.1007/s00424-018-2122-3. Epub 2018 Mar 2. Pflugers Arch. 2018. PMID: 29500669 Free PMC article.
-
Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure.Circ Res. 2018 Jul 20;123(3):356-371. doi: 10.1161/CIRCRESAHA.118.312708. Epub 2018 Jun 13. Circ Res. 2018. PMID: 29898892 Free PMC article.
References
-
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. - PubMed
-
- Nakamura R, Egashira K, Machida Y, Hayashidani S, Takeya M, Utsumi H, Tsutsui H, Takeshita A. Probucol attenuates left ventricular dysfunction and remodeling in tachycardia-induced heart failure: roles of oxidative stress and inflammation. Circulation. 2002;106:362–367. - PubMed
-
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. - PubMed
-
- Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999;85:357–363. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
