

Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice
- PMID: 20347818
- PMCID: PMC4631262
- DOI: 10.1053/j.gastro.2010.03.052
Toll-like receptor 9 promotes steatohepatitis by induction of interleukin-1beta in mice
Abstract
Background & aims: Development of nonalcoholic steatohepatitis (NASH) involves the innate immune system and is mediated by Kupffer cells and hepatic stellate cells (HSCs). Toll-like receptor 9 (TLR9) is a pattern recognition receptor that recognizes bacteria-derived cytosine phosphate guanine (CpG)-containing DNA and activates innate immunity. We investigated the role of TLR9 signaling and the inflammatory cytokine interleukin-1beta (IL-1beta) in steatohepatitis, fibrosis, and insulin resistance.
Methods: Wild-type (WT), TLR9(-/-), IL-1 receptor (IL-1R)(-/-), and MyD88(-/-) mice were fed a choline-deficient amino acid-defined (CDAA) diet for 22 weeks and then assessed for steatohepatitis, fibrosis, and insulin resistance. Lipid accumulation and cell death were assessed in isolated hepatocytes. Kupffer cells and HSCs were isolated to assess inflammatory and fibrogenic responses, respectively.
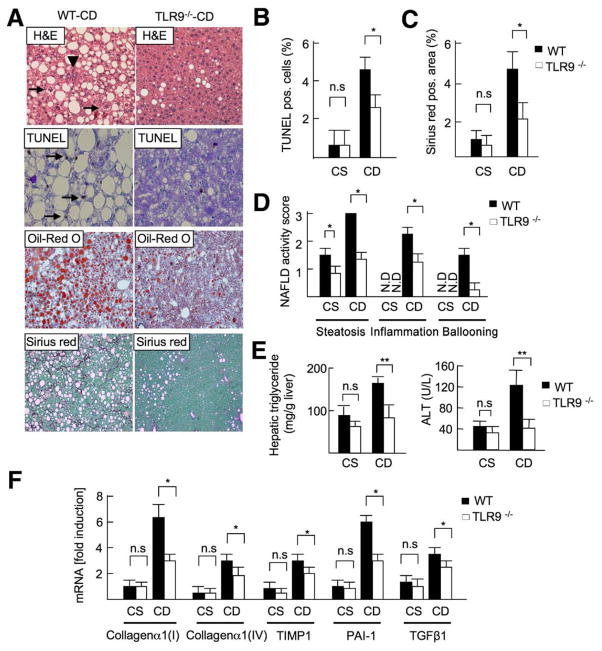
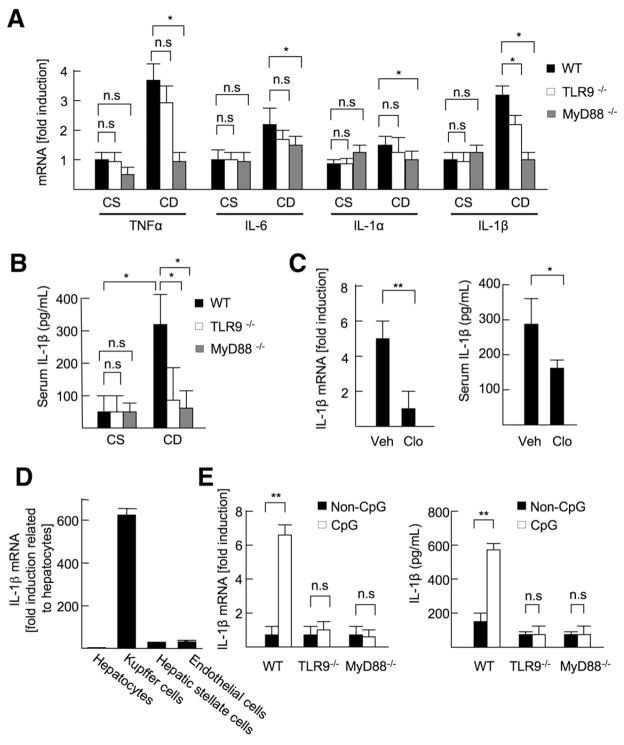
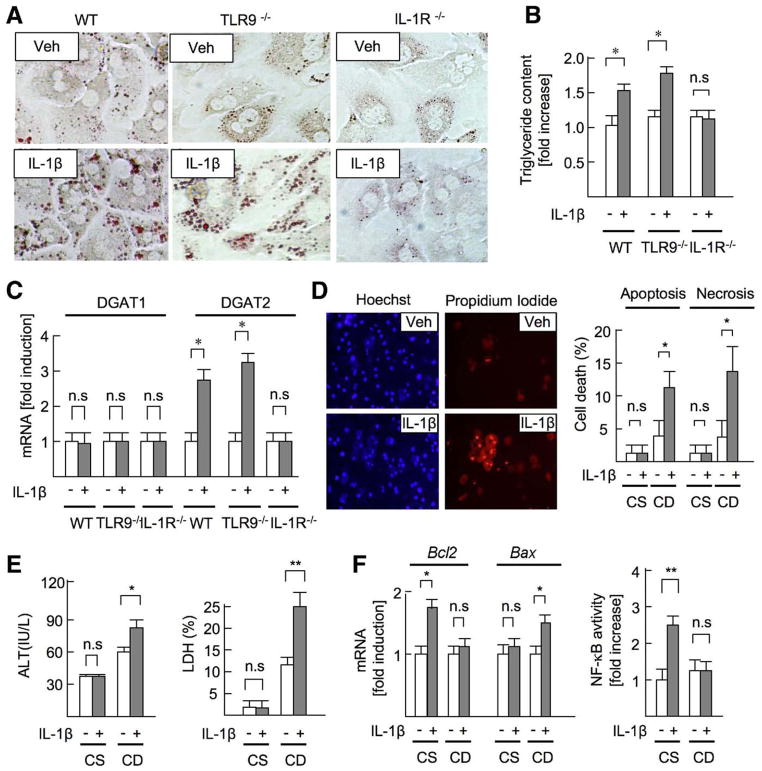
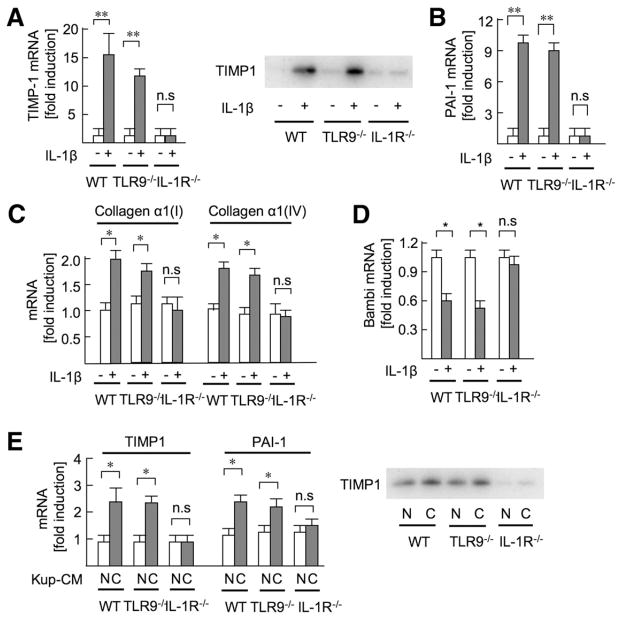
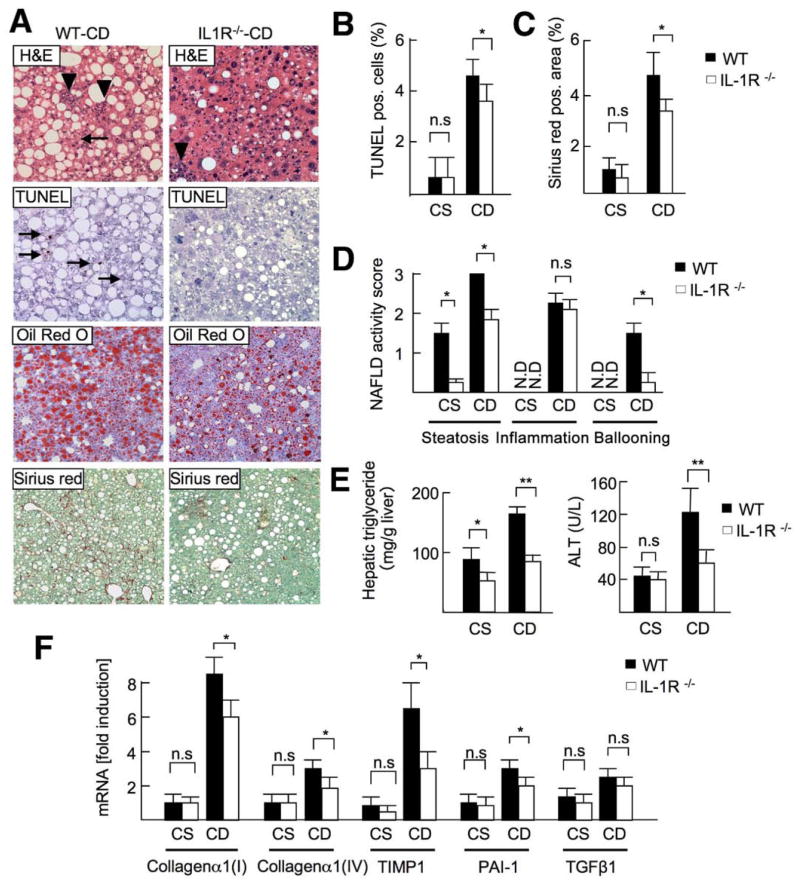
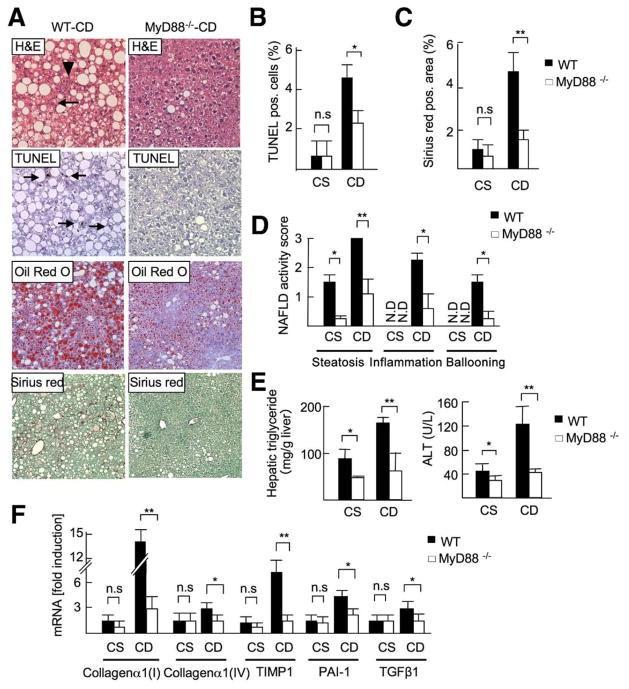
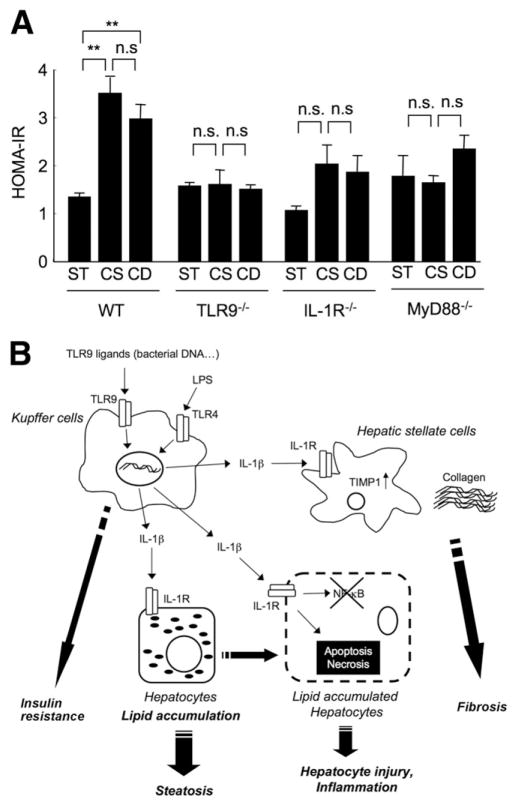
Results: The CDAA diet induced NASH in WT mice, characterized by steatosis, inflammation, fibrosis, and insulin resistance. TLR9(-/-) mice showed less steatohepatitis and liver fibrosis than WT mice. Among inflammatory cytokines, IL-1beta production was suppressed in TLR9(-/-) mice. Kupffer cells produced IL-1beta in response to CpG oligodeoxynucleotide. IL-1beta but not CpG-oligodeoxynucleotides, increased lipid accumulation in hepatocytes. Lipid accumulation in hepatocytes led to nuclear factor-kappaB inactivation, resulting in cell death in response to IL-1beta. IL-1beta induced fibrogenic responses in HSCs, including secretion of tissue inhibitor of metalloproteinase-1. IL-1R(-/-) mice had reduced steatohepatitis and fibrosis, compared with WT mice. Mice deficient in MyD88, an adaptor molecule for TLR9 and IL-1R signaling, also had reduced steatohepatitis and fibrosis. TLR9(-/-), IL-1R(-/-), and MyD88(-/-) mice had less insulin resistance than WT mice on the CDAA diet.
Conclusions: In a mouse model of NASH, TLR9 signaling induces production of IL-1beta by Kupffer cells, leading to steatosis, inflammation, and fibrosis.
Copyright 2010 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
The authors disclose no conflicts.
Figures
Comment in
-
Innate immunity and steatohepatitis: a critical role of another toll (TLR-9).Gastroenterology. 2010 Jul;139(1):27-30. doi: 10.1053/j.gastro.2010.05.018. Gastroenterology. 2010. PMID: 20639084 Free PMC article. No abstract available.
Similar articles
-
Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice.Hepatology. 2013 Feb;57(2):577-89. doi: 10.1002/hep.26081. Hepatology. 2013. PMID: 22987396 Free PMC article.
-
Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis.Liver Int. 2014 Oct;34(9):1402-13. doi: 10.1111/liv.12537. Epub 2014 Apr 17. Liver Int. 2014. PMID: 24650018 Free PMC article.
-
c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice.Gastroenterology. 2009 Oct;137(4):1467-1477.e5. doi: 10.1053/j.gastro.2009.06.045. Epub 2009 Jun 21. Gastroenterology. 2009. PMID: 19549522 Free PMC article.
-
Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis.J Gastroenterol Hepatol. 2013 Aug;28 Suppl 1(0 1):38-42. doi: 10.1111/jgh.12019. J Gastroenterol Hepatol. 2013. PMID: 23855294 Free PMC article. Review.
-
IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice.J Clin Invest. 2012 Oct;122(10):3476-89. doi: 10.1172/JCI60777. Epub 2012 Sep 4. J Clin Invest. 2012. PMID: 22945633 Free PMC article. Review.
Cited by
-
Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis.PLoS One. 2013;8(2):e56100. doi: 10.1371/journal.pone.0056100. Epub 2013 Feb 7. PLoS One. 2013. PMID: 23409132 Free PMC article.
-
Oxidant therapy improves adipogenic differentiation of adipose-derived stem cells in human wound healing.Stem Cell Res Ther. 2021 May 10;12(1):280. doi: 10.1186/s13287-021-02336-3. Stem Cell Res Ther. 2021. PMID: 33971957 Free PMC article.
-
TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration.Am J Physiol Gastrointest Liver Physiol. 2015 Jul 1;309(1):G30-41. doi: 10.1152/ajpgi.00031.2015. Epub 2015 Apr 30. Am J Physiol Gastrointest Liver Physiol. 2015. PMID: 25930080 Free PMC article.
-
Sterile inflammation in the liver and pancreas.J Gastroenterol Hepatol. 2013 Aug;28 Suppl 1(0 1):61-7. doi: 10.1111/jgh.12018. J Gastroenterol Hepatol. 2013. PMID: 23855298 Free PMC article. Review.
-
Relationship of NLRP3 inflammasome with periodontal, endodontic and related systemic diseases.Mol Biol Rep. 2022 Nov;49(11):11123-11132. doi: 10.1007/s11033-022-07894-0. Epub 2022 Sep 15. Mol Biol Rep. 2022. PMID: 36107371 Review.
References
-
- Marra F, Gastaldelli A, Svegliati Baroni G, et al. Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med. 2008;14:72–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
