

Linking functional decline of telomeres, mitochondria and stem cells during ageing
- PMID: 20336134
- PMCID: PMC3733214
- DOI: 10.1038/nature08982
Linking functional decline of telomeres, mitochondria and stem cells during ageing
Abstract
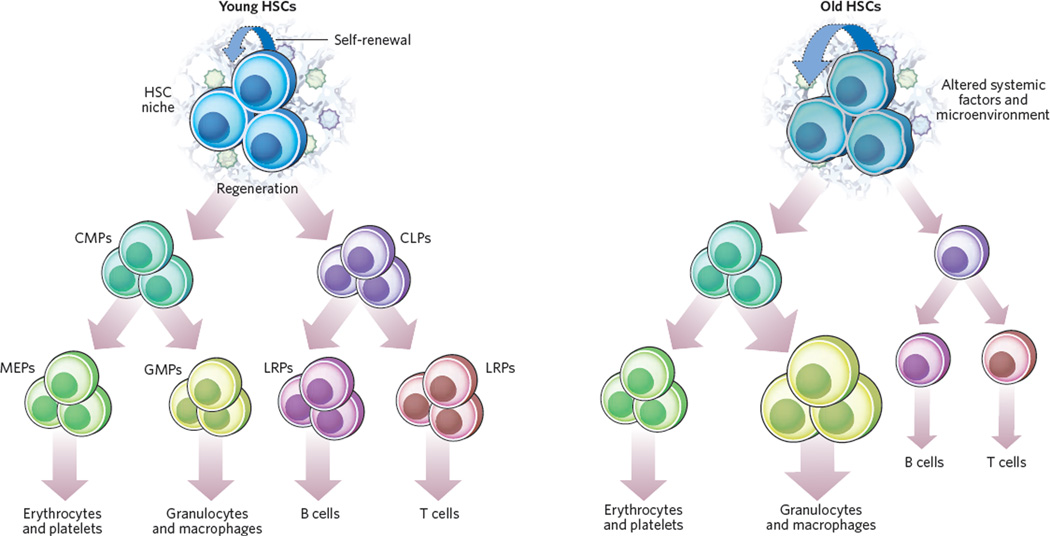
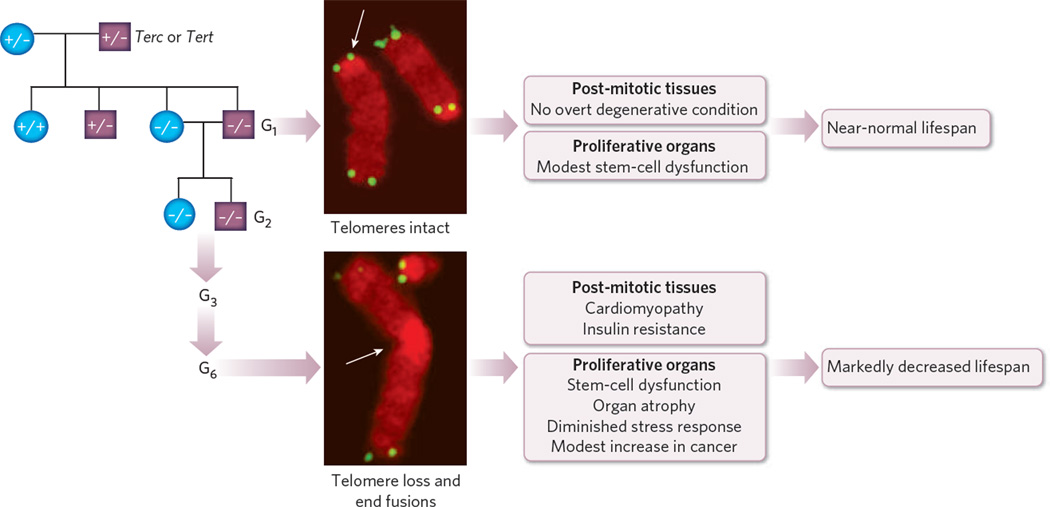
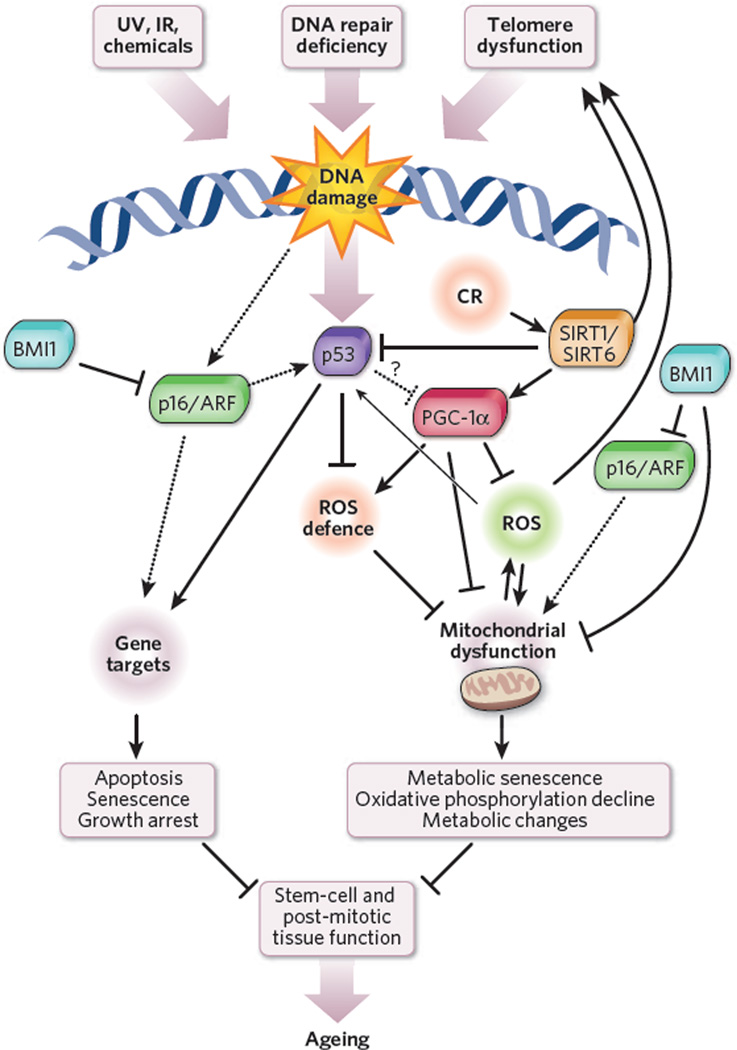
The study of human genetic disorders and mutant mouse models has provided evidence that genome maintenance mechanisms, DNA damage signalling and metabolic regulation cooperate to drive the ageing process. In particular, age-associated telomere damage, diminution of telomere 'capping' function and associated p53 activation have emerged as prime instigators of a functional decline of tissue stem cells and of mitochondrial dysfunction that adversely affect renewal and bioenergetic support in diverse tissues. Constructing a model of how telomeres, stem cells and mitochondria interact with key molecules governing genome integrity, 'stemness' and metabolism provides a framework for how diverse factors contribute to ageing and age-related disorders.
Figures
Similar articles
-
Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice.Nature. 2011 Jan 6;469(7328):102-6. doi: 10.1038/nature09603. Epub 2010 Nov 28. Nature. 2011. PMID: 21113150 Free PMC article.
-
Telomere dysfunction induces metabolic and mitochondrial compromise.Nature. 2011 Feb 17;470(7334):359-65. doi: 10.1038/nature09787. Epub 2011 Feb 9. Nature. 2011. PMID: 21307849 Free PMC article.
-
Stem cell function and maintenance - ends that matter: role of telomeres and telomerase.J Biosci. 2013 Sep;38(3):641-9. doi: 10.1007/s12038-013-9346-3. J Biosci. 2013. PMID: 23938394 Review.
-
Telomeres and mitochondria in the aging heart.Circ Res. 2012 Apr 27;110(9):1226-37. doi: 10.1161/CIRCRESAHA.111.246868. Circ Res. 2012. PMID: 22539756 Free PMC article. Review.
-
Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis.J Neurosci. 2009 Nov 18;29(46):14394-407. doi: 10.1523/JNEUROSCI.3836-09.2009. J Neurosci. 2009. PMID: 19923274 Free PMC article.
Cited by
-
GLP1 Receptor Agonists-Effects beyond Obesity and Diabetes.Cells. 2023 Dec 28;13(1):65. doi: 10.3390/cells13010065. Cells. 2023. PMID: 38201269 Free PMC article. Review.
-
Aging, Cancer, and Inflammation: The Telomerase Connection.Int J Mol Sci. 2024 Aug 5;25(15):8542. doi: 10.3390/ijms25158542. Int J Mol Sci. 2024. PMID: 39126110 Free PMC article. Review.
-
The melanocyte lineage in development and disease.Development. 2015 Feb 15;142(4):620-32. doi: 10.1242/dev.106567. Development. 2015. PMID: 25670789 Free PMC article. Review.
-
Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells.Dis Model Mech. 2016 Jul 1;9(7):719-35. doi: 10.1242/dmm.024711. Dis Model Mech. 2016. PMID: 27482812 Free PMC article. Review.
-
Telomerase downregulation induces proapoptotic genes expression and initializes breast cancer cells apoptosis followed by DNA fragmentation in a cell type dependent manner.Mol Biol Rep. 2013 Aug;40(8):4995-5004. doi: 10.1007/s11033-013-2600-9. Epub 2013 May 16. Mol Biol Rep. 2013. PMID: 23677713 Free PMC article.
References
-
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. - PubMed
-
- Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. - PubMed
-
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. - PubMed
-
- Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
