

Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex
- PMID: 20335458
- PMCID: PMC2852190
- DOI: 10.1523/JNEUROSCI.6248-09.2010
Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex
Abstract
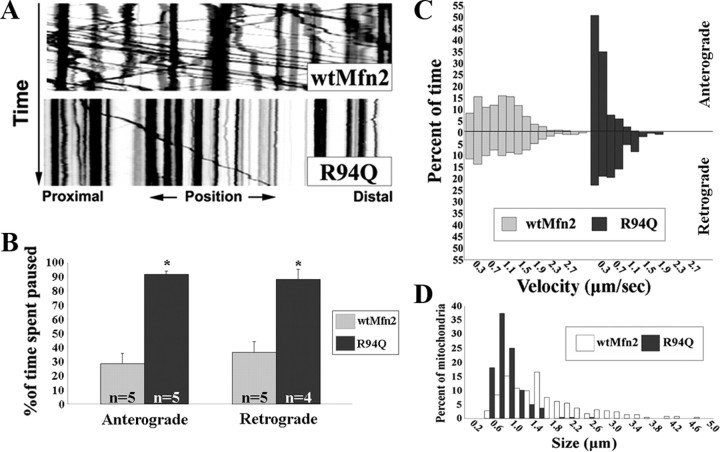
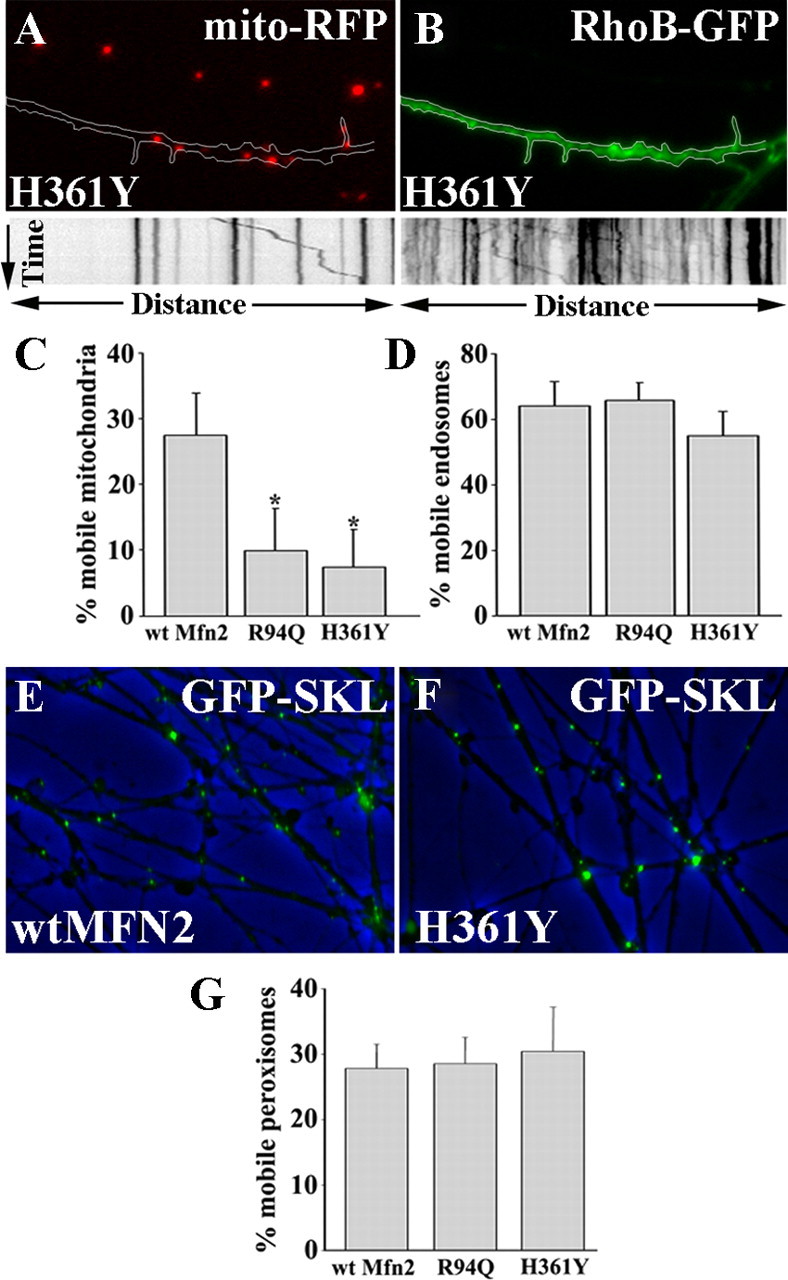
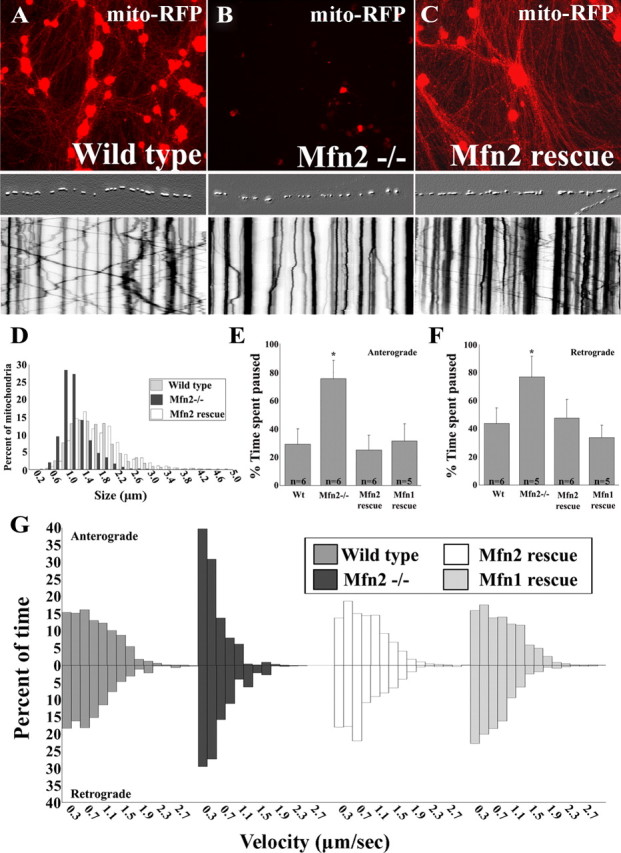
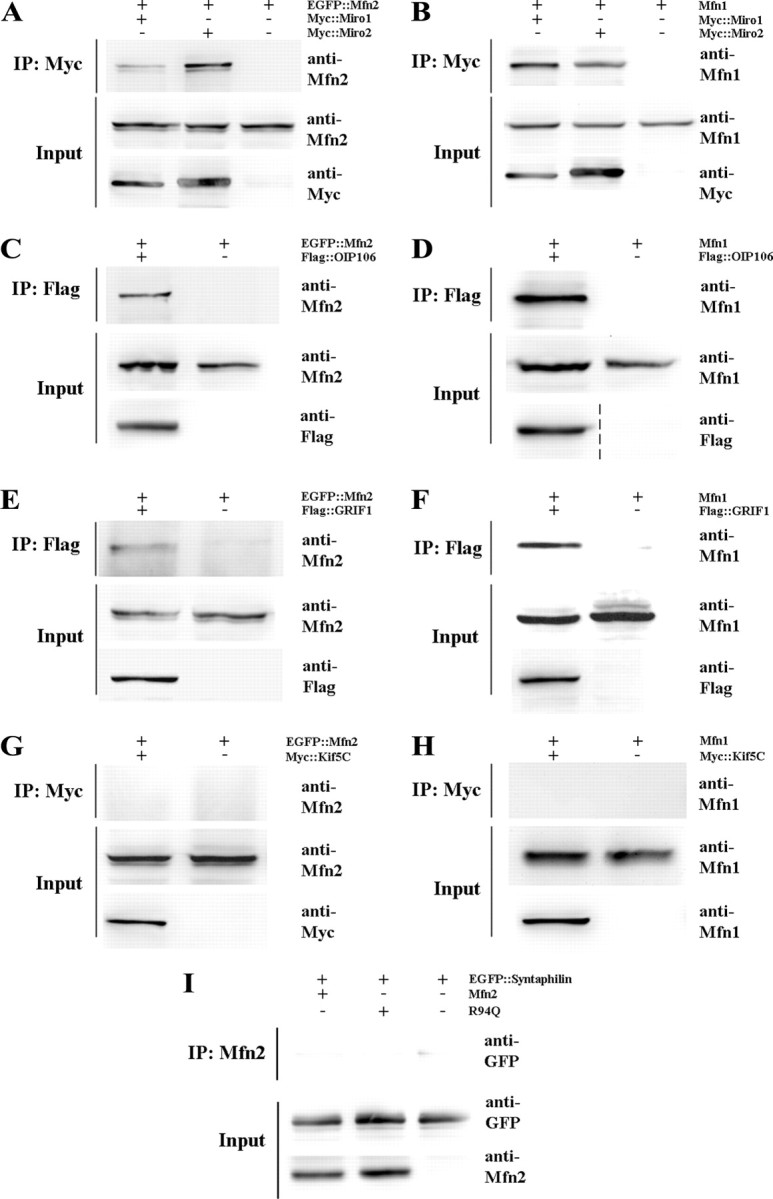
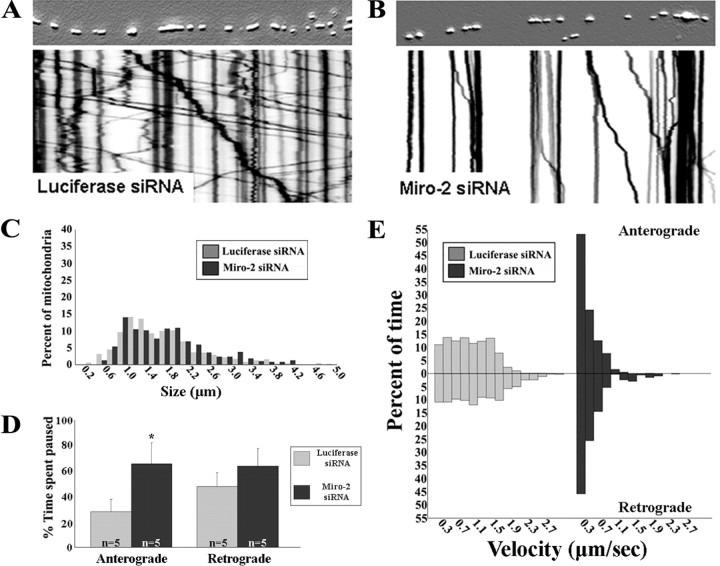
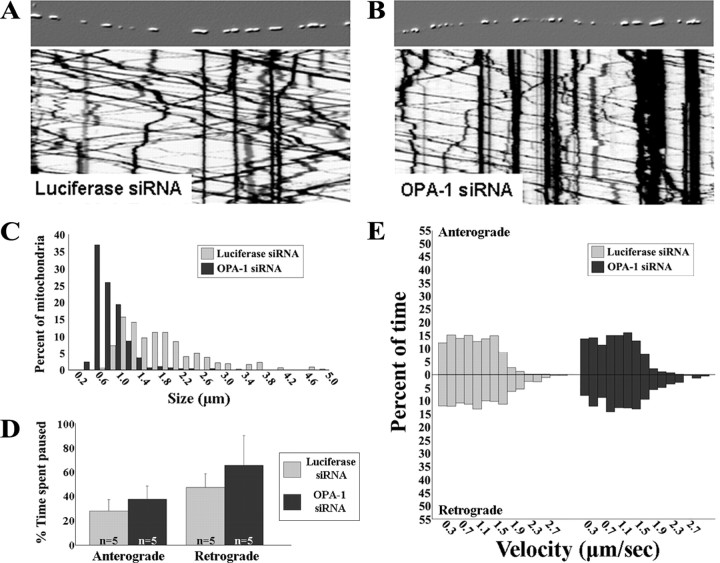
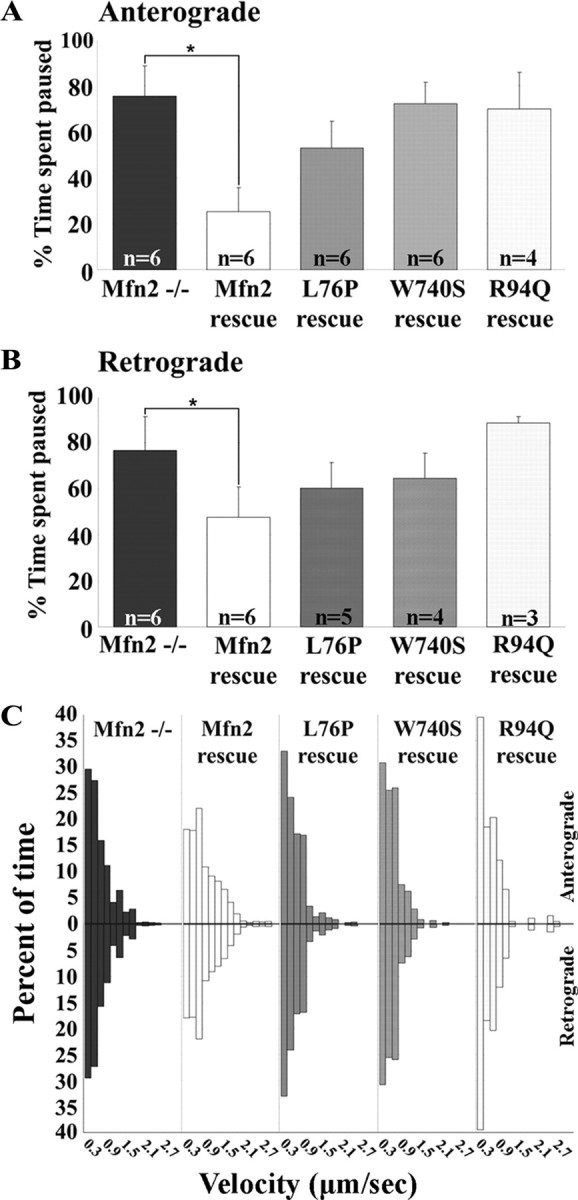
Mitofusins (Mfn1 and Mfn2) are outer mitochondrial membrane proteins involved in regulating mitochondrial dynamics. Mutations in Mfn2 cause Charcot-Marie-Tooth disease (CMT) type 2A, an inherited disease characterized by degeneration of long peripheral axons, but the nature of this tissue selectivity remains unknown. Here, we present evidence that Mfn2 is directly involved in and required for axonal mitochondrial transport, distinct from its role in mitochondrial fusion. Live imaging of neurons cultured from Mfn2 knock-out mice or neurons expressing Mfn2 disease mutants shows that axonal mitochondria spend more time paused and undergo slower anterograde and retrograde movements, indicating an alteration in attachment to microtubule-based transport systems. Furthermore, Mfn2 disruption altered mitochondrial movement selectively, leaving transport of other organelles intact. Importantly, both Mfn1 and Mfn2 interact with mammalian Miro (Miro1/Miro2) and Milton (OIP106/GRIF1) proteins, members of the molecular complex that links mitochondria to kinesin motors. Knockdown of Miro2 in cultured neurons produced transport deficits identical to loss of Mfn2, indicating that both proteins must be present at the outer membrane to mediate axonal mitochondrial transport. In contrast, disruption of mitochondrial fusion via knockdown of the inner mitochondrial membrane protein Opa1 had no effect on mitochondrial motility, indicating that loss of fusion does not inherently alter mitochondrial transport. These experiments identify a role for mitofusins in directly regulating mitochondrial transport and offer important insight into the cell type specificity and molecular mechanisms of axonal degeneration in CMT2A and dominant optic atrophy.
Figures
Similar articles
-
Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration.J Neurosci. 2012 Mar 21;32(12):4145-55. doi: 10.1523/JNEUROSCI.6338-11.2012. J Neurosci. 2012. PMID: 22442078 Free PMC article.
-
Mitochondrial dynamics and inherited peripheral nerve diseases.Neurosci Lett. 2015 Jun 2;596:66-77. doi: 10.1016/j.neulet.2015.04.001. Epub 2015 Apr 3. Neurosci Lett. 2015. PMID: 25847151 Review.
-
Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations.J Neurosci. 2007 Jan 10;27(2):422-30. doi: 10.1523/JNEUROSCI.4798-06.2007. J Neurosci. 2007. PMID: 17215403 Free PMC article.
-
Burst mitofusin activation reverses neuromuscular dysfunction in murine CMT2A.Elife. 2020 Oct 19;9:e61119. doi: 10.7554/eLife.61119. Elife. 2020. PMID: 33074106 Free PMC article.
-
Role of mitofusin 2 mutations in the physiopathology of Charcot-Marie-Tooth disease type 2A.Exp Neurol. 2009 Aug;218(2):268-73. doi: 10.1016/j.expneurol.2009.05.003. Epub 2009 May 8. Exp Neurol. 2009. PMID: 19427854 Review.
Cited by
-
Regulation of adrenocortical steroid hormone production by RhoA-diaphanous 1 signaling and the cytoskeleton.Mol Cell Endocrinol. 2013 May 22;371(1-2):79-86. doi: 10.1016/j.mce.2012.11.014. Epub 2012 Nov 24. Mol Cell Endocrinol. 2013. PMID: 23186810 Free PMC article. Review.
-
Miro1 depletion disrupts spatial distribution of mitochondria and leads to oocyte maturation defects.Front Cell Dev Biol. 2022 Oct 17;10:986454. doi: 10.3389/fcell.2022.986454. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36325364 Free PMC article.
-
Mitochondrial dynamics involves molecular and mechanical events in motility, fusion and fission.Front Cell Dev Biol. 2022 Oct 19;10:1010232. doi: 10.3389/fcell.2022.1010232. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36340034 Free PMC article. Review.
-
Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart.Circ Res. 2012 Sep 28;111(8):1012-26. doi: 10.1161/CIRCRESAHA.112.274142. Epub 2012 Aug 17. Circ Res. 2012. PMID: 22904094 Free PMC article.
-
Mitochondrial dysfunction of induced pluripotent stem cells-based neurodegenerative disease modeling and therapeutic strategy.Front Cell Dev Biol. 2022 Nov 21;10:1030390. doi: 10.3389/fcell.2022.1030390. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36478742 Free PMC article. Review.
References
-
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet. 2000;26:211–215. - PubMed
-
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. - PubMed
-
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. - PubMed
-
- Berthold CH, Fabricius C, Rydmark M, Andersen B. Axoplasmic organelles at nodes of Ranvier. I. Occurrence and distribution in large myelinated spinal root axons of the adult cat. J Neurocytol. 1993;22:925–940. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases