

Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo
- PMID: 20335454
- PMCID: PMC2855655
- DOI: 10.1523/JNEUROSCI.6393-09.2010
Deletion of M1 muscarinic acetylcholine receptors increases amyloid pathology in vitro and in vivo
Abstract
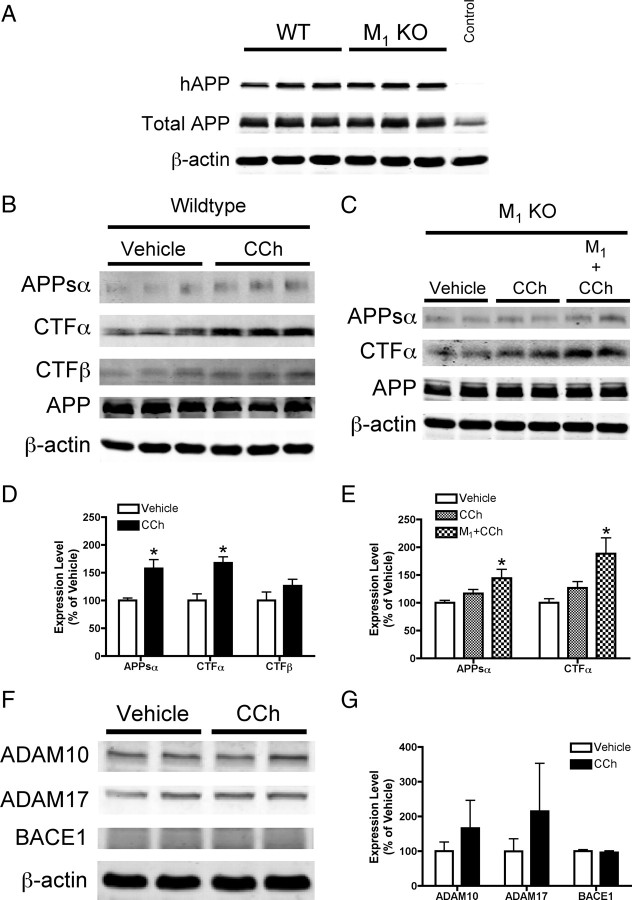
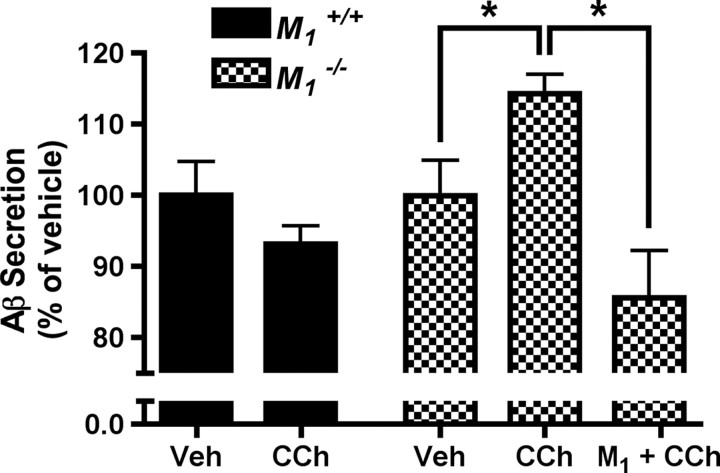
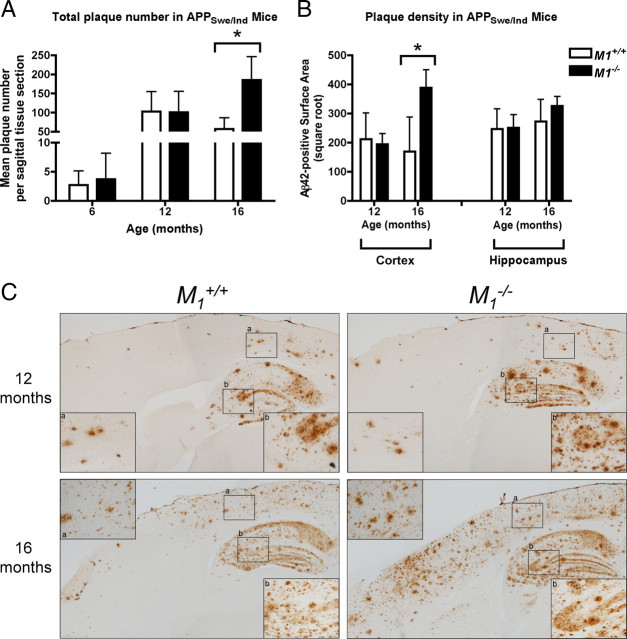
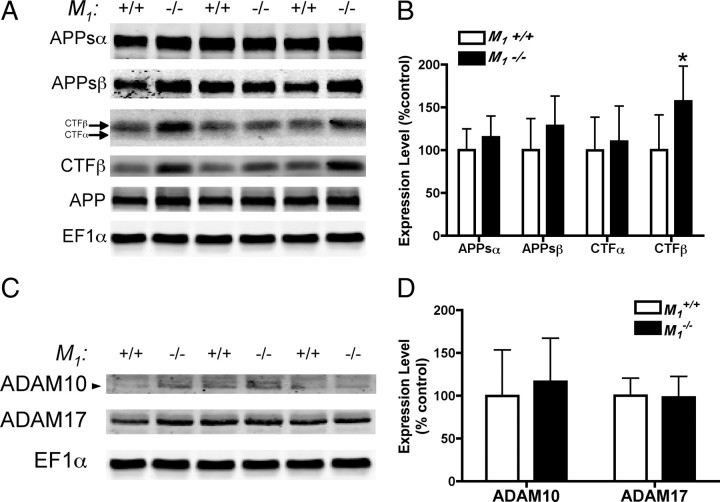
Alzheimer's disease (AD) is a progressive neurological disorder that causes dementia and poses a major public health crisis as the population ages. Aberrant processing of the amyloid precursor protein (APP) is strongly implicated as a proximal event in AD pathophysiology, but the neurochemical signals that regulate APP processing in the brain are not completely understood. Activation of muscarinic acetylcholine receptors (mAChRs) has been shown to affect APP processing and AD pathology, but less is known about the roles of specific mAChR subtypes. In this study, we used M(1) mAChR knock-out mice (M(1)KO) to isolate the effects of the M(1) mAChR on APP processing in primary neurons and on the development of amyloid pathology in a transgenic mouse model of AD. We demonstrate that the loss of M(1) mAChRs increases amyloidogenic APP processing in neurons, as evidenced by decreased agonist-regulated shedding of the neuroprotective APP ectodomain APPsalpha and increased production of toxic Abeta peptides. Expression of M(1) mAChRs on the M(1)KO background rescued this phenotype, indicating that M(1) mAChRs are sufficient to modulate nonamyloidogenic APP processing. In APP(Swe/Ind) transgenic mice, the loss of M(1) mAChRs resulted in increased levels of brain Abeta and greater accumulation of amyloid plaque pathology. Analysis of APP metabolites in APP(Swe/Ind) brain tissue indicates that the loss of M(1) mAChRs increases amyloidogenic APP processing. These results indicate that the M(1) mAChR is an important regulator of amyloidogenesis in the brain and provide strong support for targeting the M(1) mAChR as a therapeutic candidate in AD.
Figures
Similar articles
-
The metalloprotease inhibitor TIMP-3 regulates amyloid precursor protein and apolipoprotein E receptor proteolysis.J Neurosci. 2007 Oct 3;27(40):10895-905. doi: 10.1523/JNEUROSCI.3135-07.2007. J Neurosci. 2007. PMID: 17913923 Free PMC article.
-
Acute Effects of Muscarinic M1 Receptor Modulation on AβPP Metabolism and Amyloid-β Levels in vivo: A Microdialysis Study.J Alzheimers Dis. 2015;46(4):971-82. doi: 10.3233/JAD-150152. J Alzheimers Dis. 2015. PMID: 25881909
-
Deletion of Mint proteins decreases amyloid production in transgenic mouse models of Alzheimer's disease.J Neurosci. 2008 Dec 31;28(53):14392-400. doi: 10.1523/JNEUROSCI.2481-08.2008. J Neurosci. 2008. PMID: 19118172 Free PMC article.
-
The regulation of amyloid precursor protein metabolism by cholinergic mechanisms and neurotrophin receptor signaling.Prog Neurobiol. 1998 Dec;56(5):541-69. doi: 10.1016/s0301-0082(98)00044-6. Prog Neurobiol. 1998. PMID: 9775403 Review.
-
Therapeutic approaches to Alzheimer's disease through stimulating of non-amyloidogenic processing of amyloid precursor protein.Eur Rev Med Pharmacol Sci. 2016 Jun;20(11):2389-403. Eur Rev Med Pharmacol Sci. 2016. PMID: 27338066 Review.
Cited by
-
Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer's disease and schizophrenia.Neuropsychiatr Dis Treat. 2014 Jan 28;10:183-91. doi: 10.2147/NDT.S55104. eCollection 2014. Neuropsychiatr Dis Treat. 2014. PMID: 24511233 Free PMC article. Review.
-
A comprehensive assessment of the cholinergic-supporting and cognitive-enhancing effects of Rosa damascena Mill. (Damask rose) essential oil on scopolamine-induced amnestic rats.Brain Behav. 2024 May;14(5):e3507. doi: 10.1002/brb3.3507. Brain Behav. 2024. PMID: 38688895 Free PMC article.
-
Therapeutic Potential of Multifunctional Derivatives of Cholinesterase Inhibitors.Curr Neuropharmacol. 2021;19(8):1323-1344. doi: 10.2174/1570159X19666201218103434. Curr Neuropharmacol. 2021. PMID: 33342413 Free PMC article.
-
Effects of sub-chronic donepezil on brain Abeta and cognition in a mouse model of Alzheimer's disease.Psychopharmacology (Berl). 2013 Nov;230(2):279-89. doi: 10.1007/s00213-013-3152-3. Epub 2013 Jun 20. Psychopharmacology (Berl). 2013. PMID: 23783773
-
Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer's disease.Acta Neuropathol. 2011 Jul;122(1):49-60. doi: 10.1007/s00401-011-0831-1. Epub 2011 May 1. Acta Neuropathol. 2011. PMID: 21533854 Free PMC article.
References
-
- Alzheimer's Association. 2009 Alzheimer's disease facts and figures. Alzheimers Dement. 2009;5:234–270. - PubMed
-
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci. 2003;6:51–58. - PubMed
-
- Beach TG, Potter PE, Kuo YM, Emmerling MR, Durham RA, Webster SD, Walker DG, Sue LI, Scott S, Layne KJ, Roher AE. Cholinergic deafferentation of the rabbit cortex: a new animal model of Abeta deposition. Neurosci Lett. 2000;283:9–12. - PubMed
-
- Beach TG, Kuo YM, Schwab C, Walker DG, Roher AE. Reduction of cortical amyloid beta levels in guinea pig brain after systemic administration of physostigmine. Neurosci Lett. 2001a;310:21–24. - PubMed
-
- Beach TG, Walker DG, Potter PE, Sue LI, Fisher A. Reduction of cerebrospinal fluid amyloid beta after systemic administration of M1 muscarinic agonists. Brain Res. 2001b;905:220–223. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials