

Molecular advances in autosomal dominant polycystic kidney disease
- PMID: 20219615
- PMCID: PMC2837604
- DOI: 10.1053/j.ackd.2010.01.002
Molecular advances in autosomal dominant polycystic kidney disease
Abstract
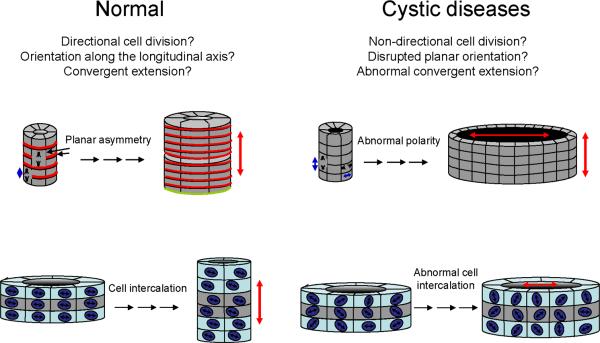
Autosomal dominant polycystic disease (ADPKD) is the most common form of inherited kidney disease that results in renal failure. The understanding of the pathogenesis of ADPKD has advanced significantly since the discovery of the 2 causative genes, PKD1 and PKD2. Dominantly inherited gene mutations followed by somatic second-hit mutations inactivating the normal copy of the respective gene result in renal tubular cyst formation that deforms the kidney and eventually impairs its function. The respective gene products, polycystin-1 and polycystin-2, work together in a common cellular pathway. Polycystin-1, a large receptor molecule, forms a receptor-channel complex with polycystin-2, which is a cation channel belonging to the TRP family. Both polycystin proteins have been localized to the primary cilium, a nonmotile microtubule-based structure that extends from the apical membrane of tubular cells into the lumen. Here we discuss recent insights in the pathogenesis of ADPKD including the genetics of ADPKD, the properties of the respective polycystin proteins, the role of cilia, and some cell-signaling pathways that have been implicated in the pathways related to PKD1 and PKD2.
2010 National Kidney Foundation, Inc. All rights reserved.
Figures
Similar articles
-
The heteromeric PC-1/PC-2 polycystin complex is activated by the PC-1 N-terminus.Elife. 2020 Nov 9;9:e60684. doi: 10.7554/eLife.60684. Elife. 2020. PMID: 33164752 Free PMC article.
-
Genetic Mechanisms of ADPKD.Adv Exp Med Biol. 2016;933:13-22. doi: 10.1007/978-981-10-2041-4_2. Adv Exp Med Biol. 2016. PMID: 27730431 Review.
-
Autosomal dominant polycystic kidney disease: recent advances in pathogenesis and potential therapies.Clin Exp Nephrol. 2013 Jun;17(3):317-26. doi: 10.1007/s10157-012-0741-0. Epub 2012 Nov 29. Clin Exp Nephrol. 2013. PMID: 23192769 Review.
-
Cilia and polycystic kidney disease.Semin Cell Dev Biol. 2021 Feb;110:139-148. doi: 10.1016/j.semcdb.2020.05.003. Epub 2020 May 28. Semin Cell Dev Biol. 2021. PMID: 32475690 Review.
-
Molecular dysregulation of ciliary polycystin-2 channels caused by variants in the TOP domain.Proc Natl Acad Sci U S A. 2020 May 12;117(19):10329-10338. doi: 10.1073/pnas.1920777117. Epub 2020 Apr 24. Proc Natl Acad Sci U S A. 2020. PMID: 32332171 Free PMC article.
Cited by
-
Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease.Vitam Horm. 2012;88:141-71. doi: 10.1016/B978-0-12-394622-5.00007-9. Vitam Horm. 2012. PMID: 22391303 Free PMC article. Review.
-
The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies.Cell Mol Life Sci. 2013 Jun;70(11):1849-74. doi: 10.1007/s00018-012-1052-z. Epub 2012 Jul 11. Cell Mol Life Sci. 2013. PMID: 22782110 Free PMC article. Review.
-
Lupus nephritis: The regulatory interplay between epigenetic and MicroRNAs.Front Physiol. 2022 Sep 16;13:925416. doi: 10.3389/fphys.2022.925416. eCollection 2022. Front Physiol. 2022. PMID: 36187762 Free PMC article. Review.
-
CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD.Cell Cycle. 2012 Nov 1;11(21):4040-6. doi: 10.4161/cc.22375. Epub 2012 Oct 3. Cell Cycle. 2012. PMID: 23032260 Free PMC article.
-
Recessive PKD1 Mutations Are Associated With Febrile Seizures and Epilepsy With Antecedent Febrile Seizures and the Genotype-Phenotype Correlation.Front Mol Neurosci. 2022 May 10;15:861159. doi: 10.3389/fnmol.2022.861159. eCollection 2022. Front Mol Neurosci. 2022. PMID: 35620448 Free PMC article.
References
-
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. - PubMed
-
- The European Polycystic Kidney Disease Consortium The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. Cell. 1994;78:725. - PubMed
-
- The International Polycystic Kidney Disease Consortium Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell. 1995;81:289–298. - PubMed
-
- Mochizuki T, Wu G, Hayashi T, Xenophontos SL, Veldhuisen B, Saris JJ, Reynolds DM, Cai Y, Gabow PA, Pierides A, et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science. 1996;272:1339–1342. - PubMed
-
- Onuchic LF, Furu L, Nagasawa Y, Hou X, Eggermann T, Ren Z, Bergmann C, Senderek J, Esquivel E, Zeltner R, et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002;70:1305–1317. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
