

Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells
- PMID: 20159619
- PMCID: PMC4048034
- DOI: 10.1016/j.chom.2010.01.005
Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing gamma/delta T cells
Abstract
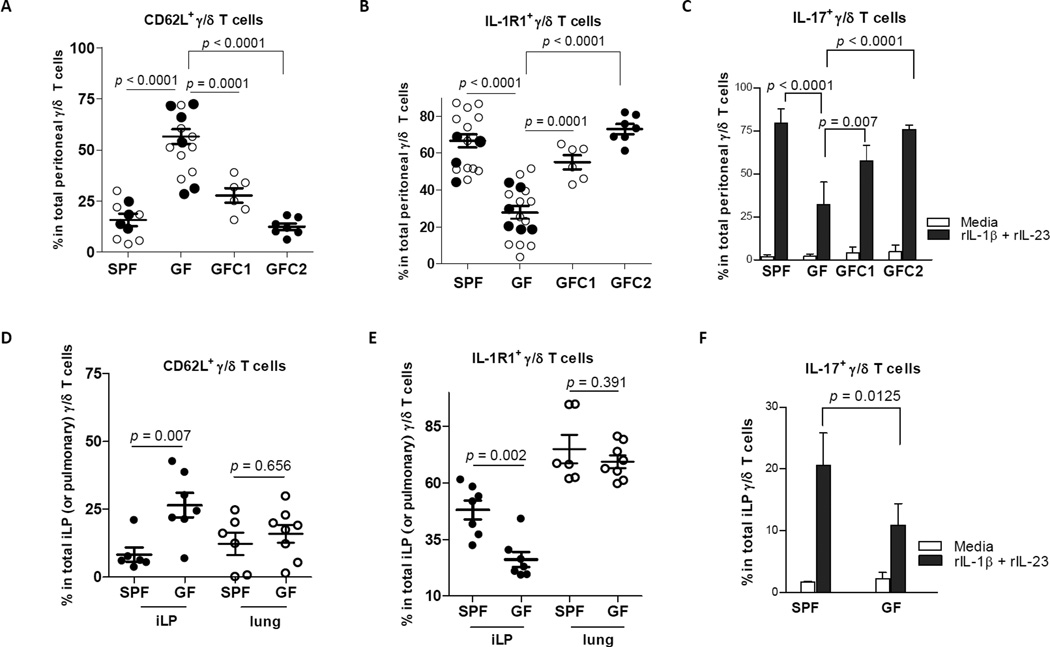
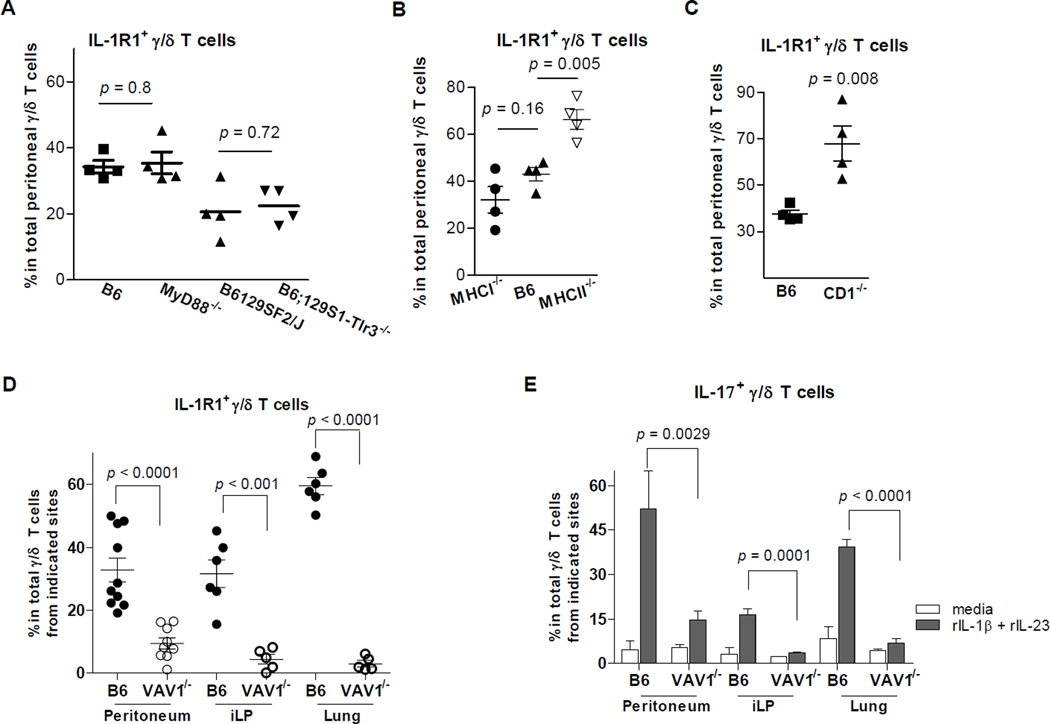
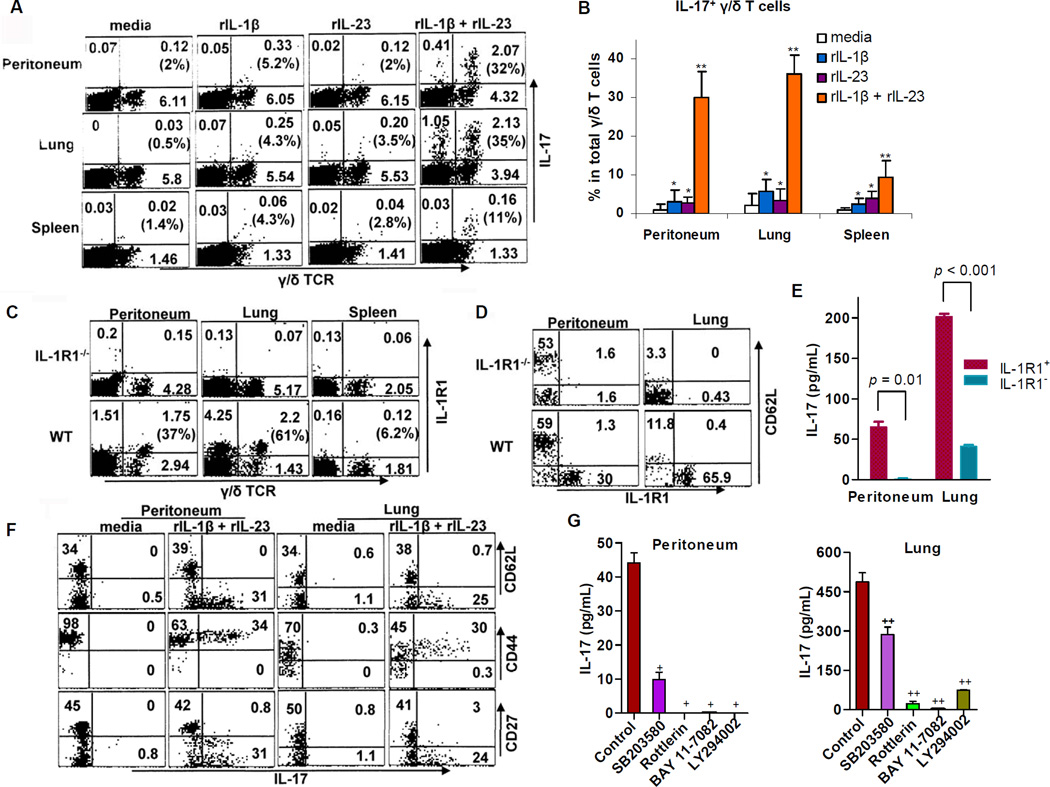
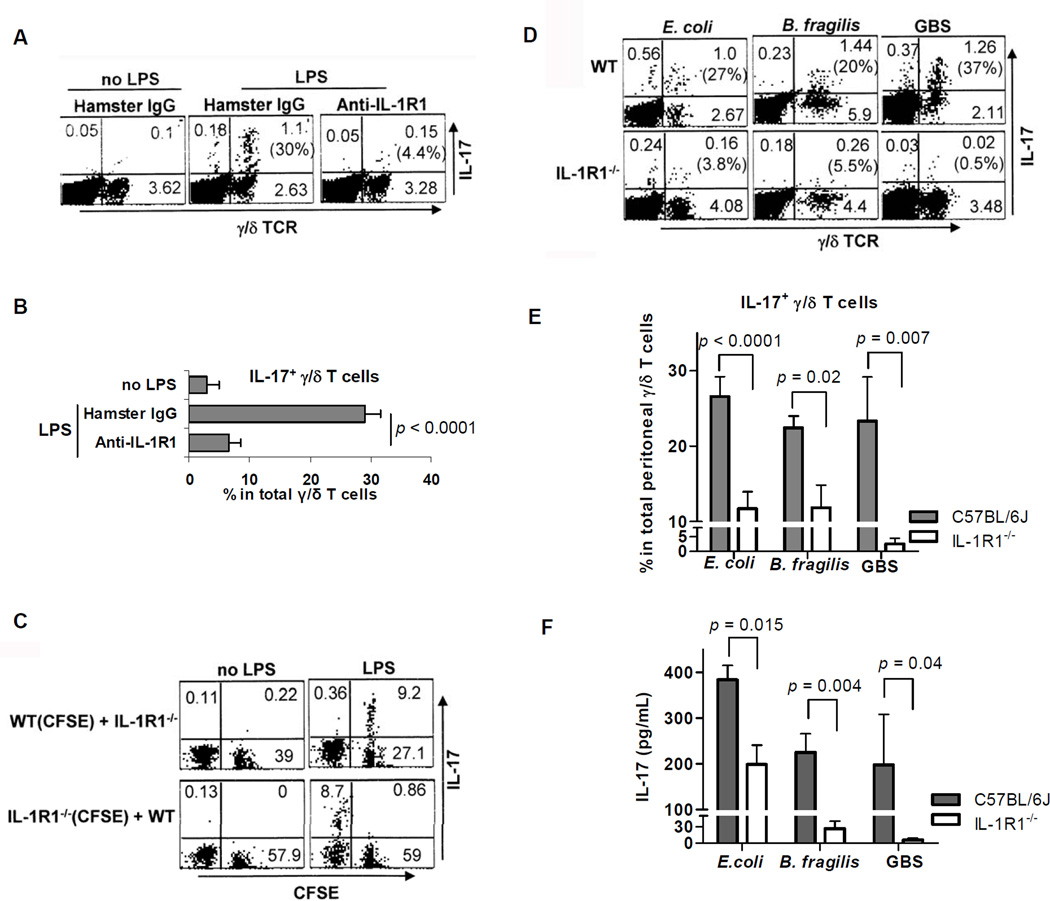
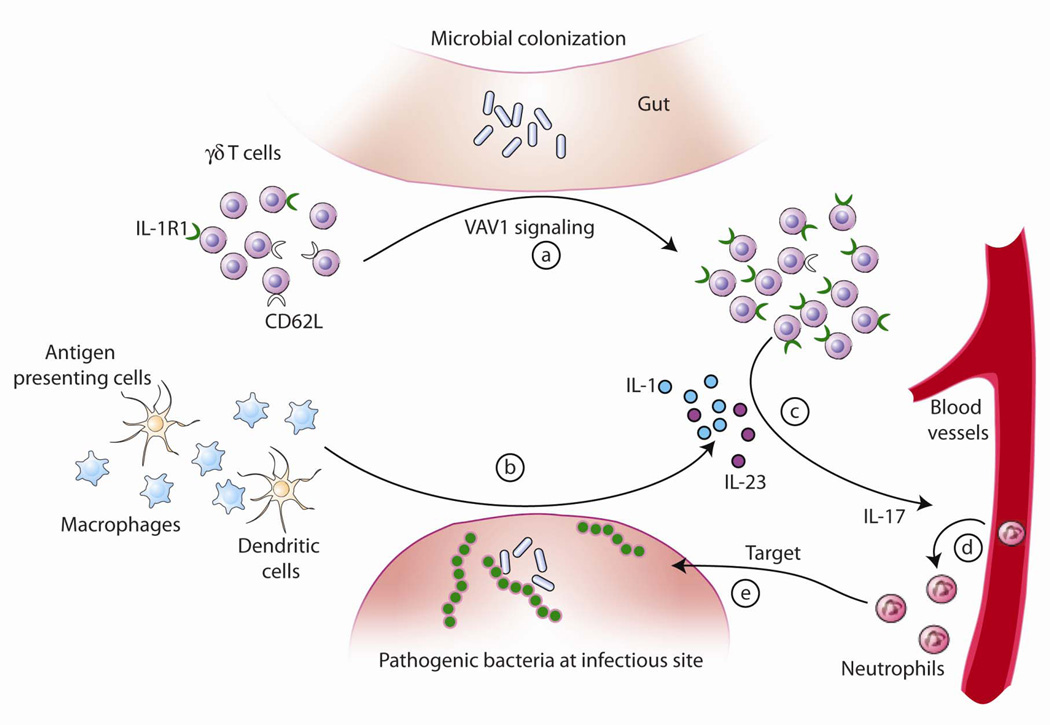
IL-17 cytokine production by the Th17 T cell subset is regulated by intestinal commmensals. We show that microbial colonization also regulates innate IL-17 production. A population of CD62L(-) gamma/delta T cells, in particular a lineage expressing the IL-1 receptor 1 (IL-1R1), can be quickly activated by microbes to produce IL-17. Antibiotic treatment and monocolonization of mice suggest that specific commensals-but not metronidazole-sensitive anaerobes like Bacteroides species-are required for maintaining IL-1R1(+) gamma/delta T cells. Signaling through the guanine nucleotide exchange factor VAV1, but not through Toll-like receptors or antigen presentation pathways, is essential for inducing IL-1R1(+) gamma/delta T cells. Furthermore, IL-1R1(+) gamma/delta T cells are a potential source of IL-17 that can be activated by IL-23 and IL-1 in both infectious and noninfectious settings in vitro and in vivo. Thus, commensals orchestrate the expansion of phenotypically distinct gammadelta T cells, and innate immunity is a three-way interaction between host, pathogens, and microbiota.
2010 Elsevier Inc. All rights reserved.
Figures

Similar articles
-
gammadelta T cells: an important source of IL-17.Curr Opin Immunol. 2008 Jun;20(3):353-7. doi: 10.1016/j.coi.2008.03.006. Epub 2008 Apr 23. Curr Opin Immunol. 2008. PMID: 18439808 Free PMC article. Review.
-
Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity.Immunity. 2009 Aug 21;31(2):331-41. doi: 10.1016/j.immuni.2009.08.001. Epub 2009 Aug 13. Immunity. 2009. PMID: 19682929
-
Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals.Immunity. 2009 Aug 21;31(2):321-30. doi: 10.1016/j.immuni.2009.06.020. Epub 2009 Aug 13. Immunity. 2009. PMID: 19682928
-
IL-17A produced by gammadelta T cells plays a critical role in innate immunity against listeria monocytogenes infection in the liver.J Immunol. 2008 Sep 1;181(5):3456-63. doi: 10.4049/jimmunol.181.5.3456. J Immunol. 2008. PMID: 18714018 Free PMC article.
-
IL-17-producing γδ T cells and innate lymphoid cells.Eur J Immunol. 2012 Sep;42(9):2221-31. doi: 10.1002/eji.201242569. Eur J Immunol. 2012. PMID: 22949320 Review.
Cited by
-
Microbes, the gut and ankylosing spondylitis.Arthritis Res Ther. 2013;15(3):214. doi: 10.1186/ar4228. Arthritis Res Ther. 2013. PMID: 23750937 Free PMC article. Review.
-
The membrane-bound mucin Muc1 regulates T helper 17-cell responses and colitis in mice.Gastroenterology. 2012 Apr;142(4):865-874.e2. doi: 10.1053/j.gastro.2011.12.036. Epub 2011 Dec 24. Gastroenterology. 2012. PMID: 22202458 Free PMC article.
-
Lung Epithelial Cells Coordinate Innate Lymphocytes and Immunity against Pulmonary Fungal Infection.Cell Host Microbe. 2018 Apr 11;23(4):511-522.e5. doi: 10.1016/j.chom.2018.02.011. Epub 2018 Mar 22. Cell Host Microbe. 2018. PMID: 29576482 Free PMC article.
-
The influence of the commensal microbiota on distal tumor-promoting inflammation.Semin Immunol. 2017 Aug;32:62-73. doi: 10.1016/j.smim.2017.06.002. Epub 2017 Jul 4. Semin Immunol. 2017. PMID: 28687194 Free PMC article. Review.
-
Sheng-Jiang powder ameliorates NAFLD via regulating intestinal microbiota in mice.Front Microbiol. 2024 May 27;15:1387401. doi: 10.3389/fmicb.2024.1387401. eCollection 2024. Front Microbiol. 2024. PMID: 38860223 Free PMC article.
References
-
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. - PubMed
-
- Akira S. Toll-like receptor signaling. J. Biol. Chem. 2003;278(40):38105–38108. - PubMed
-
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455(7214):808–812. - PubMed
-
- Chao CC, Jensen R, Dailey MO. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 1997;159(4):1686–1694. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
