

The Elk-1 and serum response factor binding sites in the major immediate-early promoter of human cytomegalovirus are required for efficient viral replication in quiescent cells and compensate for inactivation of the NF-kappaB sites in proliferating cells
- PMID: 20147408
- PMCID: PMC2863749
- DOI: 10.1128/JVI.02141-09
The Elk-1 and serum response factor binding sites in the major immediate-early promoter of human cytomegalovirus are required for efficient viral replication in quiescent cells and compensate for inactivation of the NF-kappaB sites in proliferating cells
Abstract
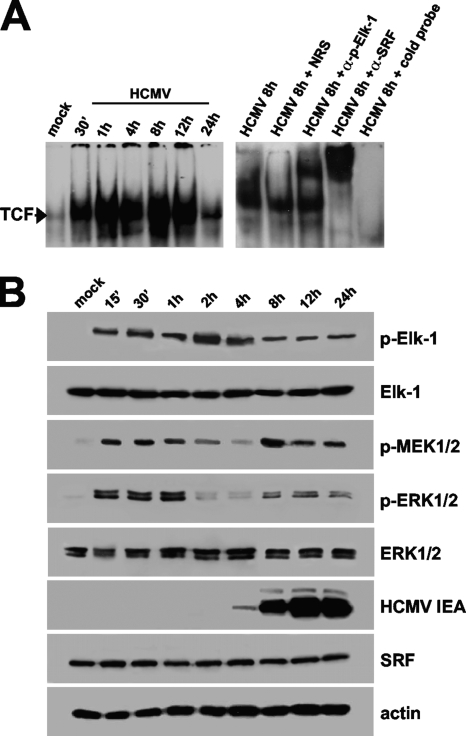
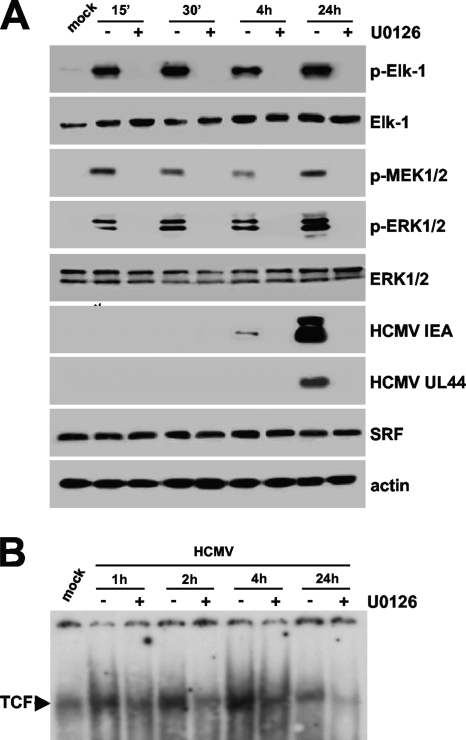
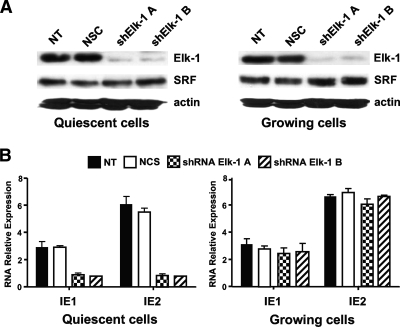
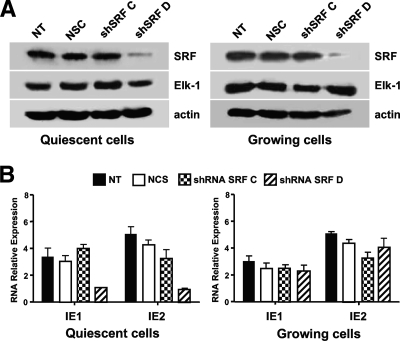
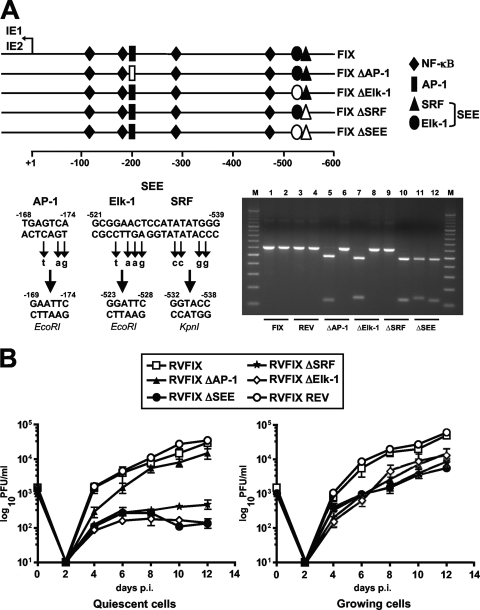
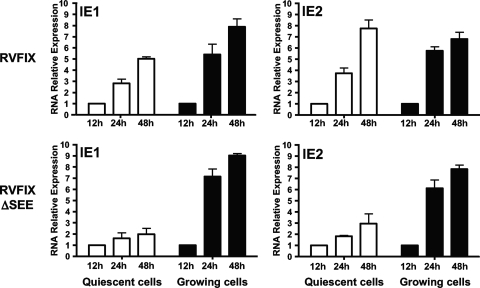
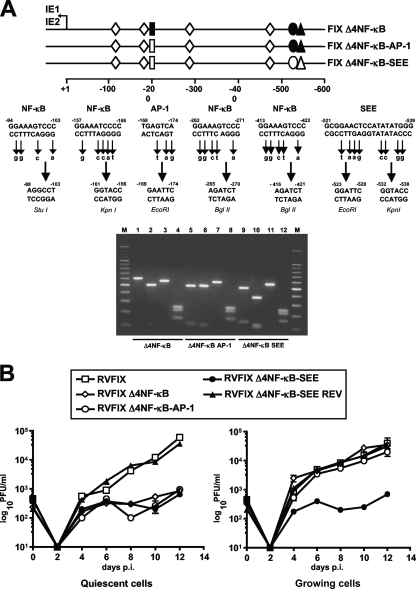
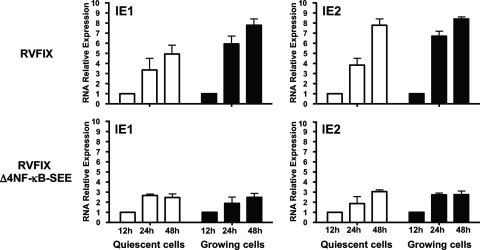
The major immediate-early promoter (MIEP) region of human cytomegalovirus (HCMV) plays a critical role in the regulation of lytic and latent infections by integrating multiple signals supplied by the infecting virus, the type and physiological state of the host cell, and its extracellular surroundings. The interaction of cellular transcription factors with their cognate binding sites, which are present at high densities within the enhancer upstream from the MIEP core promoter, regulate the rate of IE gene transcription and thus affect the outcome of HCMV infection. We have shown previously that the NF-kappaB binding sites within the MIEP enhancer and cellular NF-kappaB activity induced by HCMV infection are required for efficient MIEP activity and viral replication in quiescent cells (P. Caposio, A. Luganini, G. Hahn, S. Landolfo, and G. Gribaudo, Cell. Microbiol. 9:2040-2054, 2007). We now show that the inactivation of either the Elk-1 or serum response factor (SRF) binding site within the enhancer also reduces MIEP activation and viral replication of recombinant HCMV viruses in quiescent fibroblasts. In these cells, we show that the expression of either Elk-1 or SRF is required for optimal IE gene expression, and that the HCMV-stimulated activation of the MEK1/2-ERK1/2 signaling axis leads to Elk-1 transcriptional competency. Furthermore, the replication kinetics of recombinant viruses in which NF-kappaB, Elk-1, and SRF binding sites all are inactivated demonstrate that the higher levels of Elk-1 and SRF binding to MIEP in proliferating cells can compensate even for a lack of HCMV-induced NF-kappaB-mediated MIEP transactivation. These observations highlight the importance of the combination of different MIEP binding sites to optimize IE gene expression in cells in different physiological states.
Figures
Similar articles
-
Activation of the virus-induced IKK/NF-kappaB signalling axis is critical for the replication of human cytomegalovirus in quiescent cells.Cell Microbiol. 2007 Aug;9(8):2040-54. doi: 10.1111/j.1462-5822.2007.00936.x. Epub 2007 Apr 5. Cell Microbiol. 2007. PMID: 17419715
-
Regulation of the transcription and replication cycle of human cytomegalovirus is insensitive to genetic elimination of the cognate NF-kappaB binding sites in the enhancer.J Virol. 2006 Oct;80(19):9899-904. doi: 10.1128/JVI.00640-06. J Virol. 2006. PMID: 16973595 Free PMC article.
-
Synergistic interactions between overlapping binding sites for the serum response factor and ELK-1 proteins mediate both basal enhancement and phorbol ester responsiveness of primate cytomegalovirus major immediate-early promoters in monocyte and T-lymphocyte cell types.J Virol. 1996 Dec;70(12):8590-605. doi: 10.1128/JVI.70.12.8590-8605.1996. J Virol. 1996. PMID: 8970984 Free PMC article.
-
Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection.Biochim Biophys Acta. 2010 Mar-Apr;1799(3-4):286-95. doi: 10.1016/j.bbagrm.2009.08.001. Epub 2009 Aug 12. Biochim Biophys Acta. 2010. PMID: 19682613 Review.
-
Chromatin-mediated regulation of cytomegalovirus gene expression.Virus Res. 2011 May;157(2):134-43. doi: 10.1016/j.virusres.2010.09.019. Epub 2010 Sep 25. Virus Res. 2011. PMID: 20875471 Free PMC article. Review.
Cited by
-
Demonstration of Tightly Radiation-Controlled Molecular Switch Based on CArG Repeats by In Vivo Molecular Imaging.Mol Imaging Biol. 2015 Dec;17(6):802-10. doi: 10.1007/s11307-015-0843-7. Mol Imaging Biol. 2015. PMID: 25962973
-
The US16 gene of human cytomegalovirus is required for efficient viral infection of endothelial and epithelial cells.J Virol. 2012 Jun;86(12):6875-88. doi: 10.1128/JVI.06310-11. Epub 2012 Apr 11. J Virol. 2012. PMID: 22496217 Free PMC article.
-
Human Cytomegalovirus Latency: Approaching the Gordian Knot.Annu Rev Virol. 2016 Sep 29;3(1):333-357. doi: 10.1146/annurev-virology-110615-042422. Epub 2016 Aug 4. Annu Rev Virol. 2016. PMID: 27501258 Free PMC article.
-
A temporal gate for viral enhancers to co-opt Toll-like-receptor transcriptional activation pathways upon acute infection.PLoS Pathog. 2015 Apr 9;11(4):e1004737. doi: 10.1371/journal.ppat.1004737. eCollection 2015 Apr. PLoS Pathog. 2015. PMID: 25856589 Free PMC article.
-
Human cytomegalovirus reactivation from latency: validation of a "switch" model in vitro.Virol J. 2016 Oct 22;13(1):179. doi: 10.1186/s12985-016-0634-z. Virol J. 2016. PMID: 27770817 Free PMC article.
References
-
- Bissinger, A. L., C. Sinzger, E. Kaiserling, and G. Jahn. 2002. Human cytomegalovirus as a direct pathogen: correlation of multiorgan involvement and cell distribution with clinical and pathological findings in a case of congenital inclusion disease. J. Med. Virol. 67:200-206. - PubMed
-
- Boldogh, I., M. P. Fons, and T. Albrecht. 1993. Increased levels of sequence-specific DNA-binding proteins in human cytomegalovirus infected cells. Biochem. Biophys. Res. Commun. 197:1505-1510. - PubMed
-
- Britt, W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417-470. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
