

Neuropilin 1 directly interacts with Fer kinase to mediate semaphorin 3A-induced death of cortical neurons
- PMID: 20133938
- PMCID: PMC2843238
- DOI: 10.1074/jbc.M109.080689
Neuropilin 1 directly interacts with Fer kinase to mediate semaphorin 3A-induced death of cortical neurons
Abstract
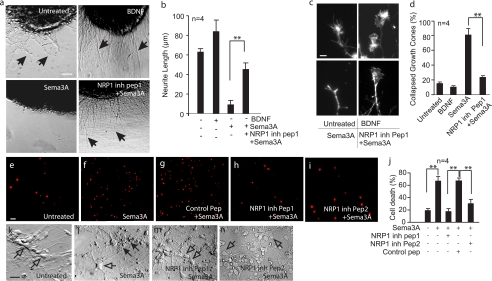
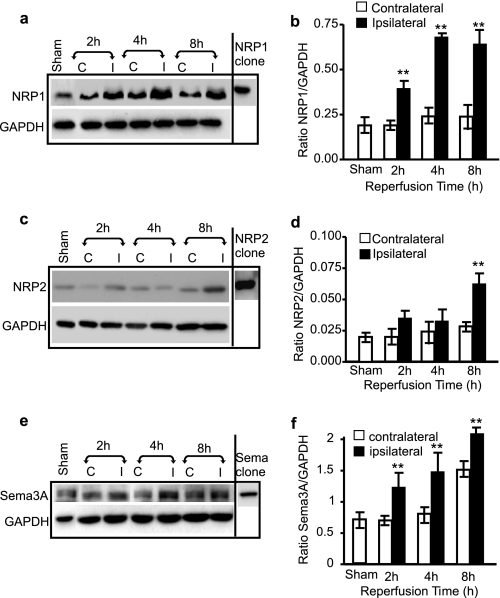
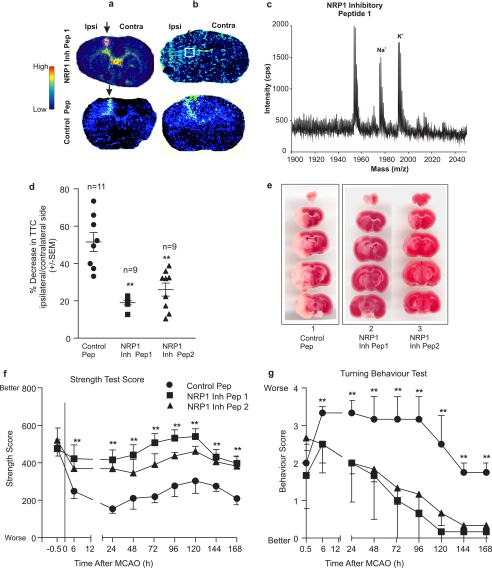
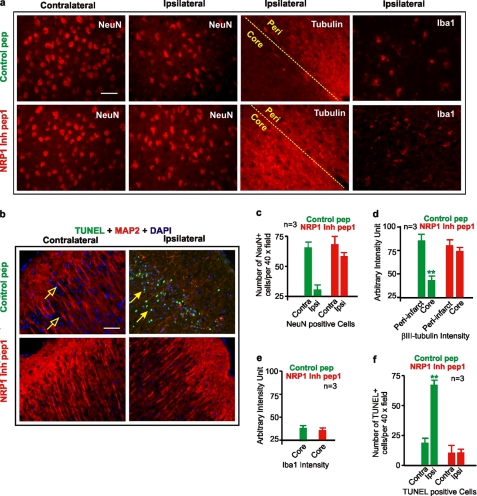
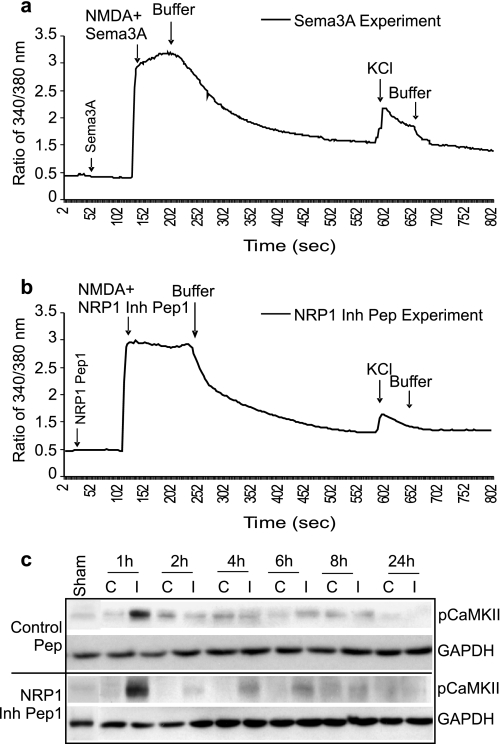
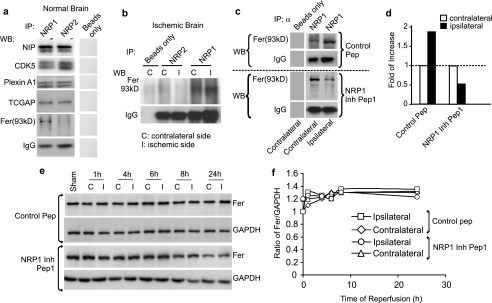
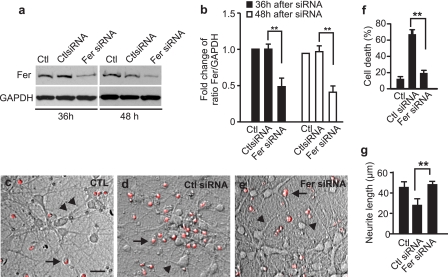
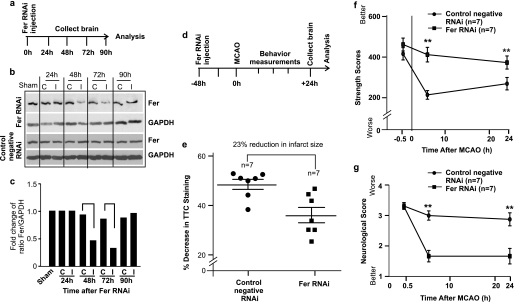
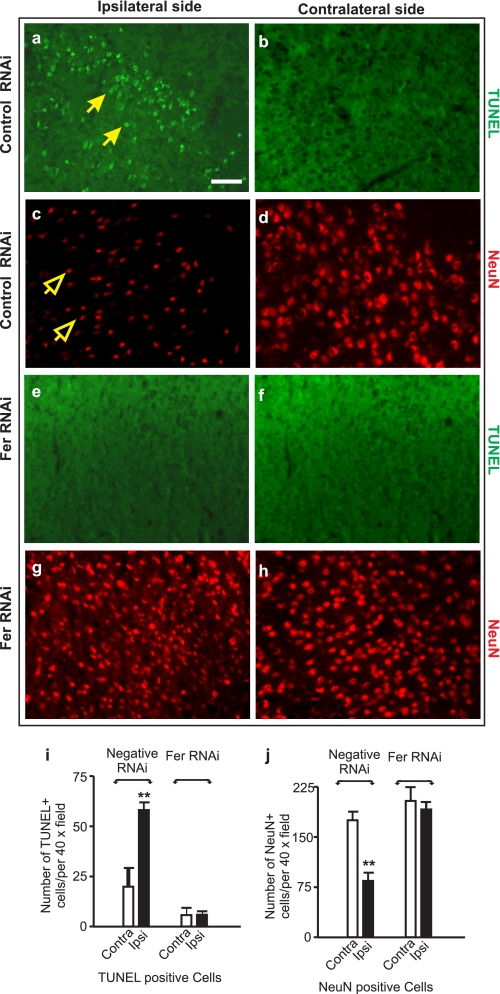
Neuropilins (NRPs) are receptors for the major chemorepulsive axonal guidance cue semaphorins (Sema). The interaction of Sema3A/NRP1 during development leads to the collapse of growth cones. Here we show that Sema3A also induces death of cultured cortical neurons through NRP1. A specific NRP1 inhibitory peptide ameliorated Sema3A-evoked cortical axonal retraction and neuronal death. Moreover, Sema3A was also involved in cerebral ischemia-induced neuronal death. Expression levels of Sema3A and NRP1, but not NRP2, were significantly increased early during brain reperfusion following transient focal cerebral ischemia. NRP1 inhibitory peptide delivered to the ischemic brain was potently neuroprotective and prevented the loss of motor functions in mice. The integrity of the injected NRP1 inhibitory peptide into the brain remained unchanged, and the intact peptide permeated the ischemic hemisphere of the brain as determined using MALDI-MS-based imaging. Mechanistically, NRP1-mediated axonal collapse and neuronal death is through direct and selective interaction with the cytoplasmic tyrosine kinase Fer. Fer RNA interference effectively attenuated Sema3A-induced neurite retraction and neuronal death in cortical neurons. More importantly, down-regulation of Fer expression using Fer-specific RNA interference attenuated cerebral ischemia-induced brain damage. Together, these studies revealed a previously unknown function of NRP1 in signaling Sema3A-evoked neuronal death through Fer in cortical neurons.
Figures
Similar articles
-
Semaphorin3A-neuropilin1 signalling is involved in the generation of cortical interneurons.Brain Struct Funct. 2017 Jul;222(5):2217-2233. doi: 10.1007/s00429-016-1337-3. Epub 2016 Nov 17. Brain Struct Funct. 2017. PMID: 27858201 Free PMC article.
-
Sustained up-regulation of semaphorin 3A, Neuropilin1, and doublecortin expression in ischemic mouse brain during long-term recovery.Biochem Biophys Res Commun. 2008 Feb 29;367(1):109-15. doi: 10.1016/j.bbrc.2007.12.103. Epub 2007 Dec 26. Biochem Biophys Res Commun. 2008. PMID: 18162177
-
Palmitoylation regulates neuropilin-2 localization and function in cortical neurons and conveys specificity to semaphorin signaling via palmitoyl acyltransferases.Elife. 2023 Apr 3;12:e83217. doi: 10.7554/eLife.83217. Elife. 2023. PMID: 37010951 Free PMC article.
-
Molecular basis of semaphorin-mediated axon guidance.J Neurobiol. 2000 Aug;44(2):219-29. doi: 10.1002/1097-4695(200008)44:2<219::aid-neu11>3.0.co;2-w. J Neurobiol. 2000. PMID: 10934324 Review.
-
The Chemorepulsive Protein Semaphorin 3A and Perineuronal Net-Mediated Plasticity.Neural Plast. 2016;2016:3679545. doi: 10.1155/2016/3679545. Epub 2016 Jan 14. Neural Plast. 2016. PMID: 27057361 Free PMC article. Review.
Cited by
-
Breast cancer cells promote osteoblastic differentiation via Sema 3A signaling pathway in vitro.Int J Clin Exp Pathol. 2015 Feb 1;8(2):1584-93. eCollection 2015. Int J Clin Exp Pathol. 2015. PMID: 25973043 Free PMC article.
-
Human airway smooth muscle cell proliferation from asthmatics is negatively regulated by semaphorin3A.Oncotarget. 2016 Dec 6;7(49):80238-80251. doi: 10.18632/oncotarget.12884. Oncotarget. 2016. PMID: 27791986 Free PMC article.
-
Collapsin response mediator protein 3 deacetylates histone H4 to mediate nuclear condensation and neuronal death.Sci Rep. 2013;3:1350. doi: 10.1038/srep01350. Sci Rep. 2013. PMID: 23443259 Free PMC article.
-
A perspective on the role of class III semaphorin signaling in central nervous system trauma.Front Cell Neurosci. 2014 Oct 27;8:328. doi: 10.3389/fncel.2014.00328. eCollection 2014. Front Cell Neurosci. 2014. PMID: 25386118 Free PMC article. Review.
-
Semaphorin 3A is a retrograde cell death signal in developing sympathetic neurons.Development. 2016 May 1;143(9):1560-70. doi: 10.1242/dev.134627. Development. 2016. PMID: 27143756 Free PMC article.
References
-
- De Winter F., Oudega M., Lankhorst A. J., Hamers F. P., Blits B., Ruitenberg M. J., Pasterkamp R. J., Gispen W. H., Verhaagen J. (2002) Exp. Neurol. 175, 61–75 - PubMed
-
- Giger R. J., Pasterkamp R. J., Holtmaat A. J., Verhaagen J. (1998) Prog. Brain Res. 117, 133–149 - PubMed
-
- Pasterkamp R. J., De Winter F., Giger R. J., Verhaagen J. (1998) Prog. Brain Res. 117, 151–170 - PubMed
-
- Pasterkamp R. J., Giger R. J., Verhaagen J. (1998) Exp. Neurol. 153, 313–327 - PubMed
-
- He Z., Koprivica V. (2004) Annu. Rev. Neurosci 27, 341–368 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
