

SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis
- PMID: 20110243
- PMCID: PMC2822627
- DOI: 10.1093/brain/awp325
SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis
Abstract
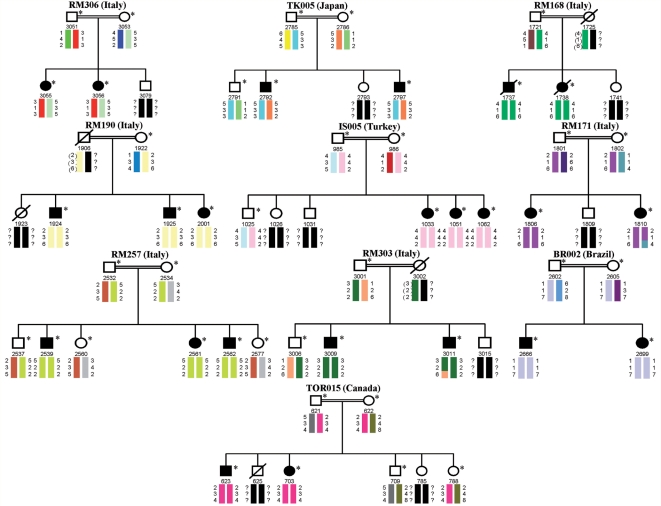
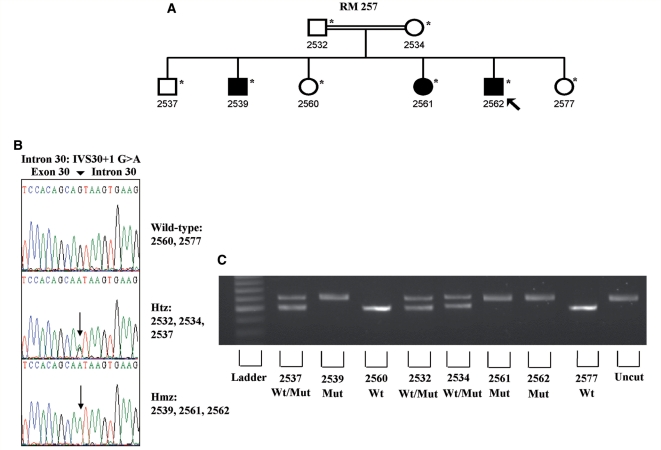
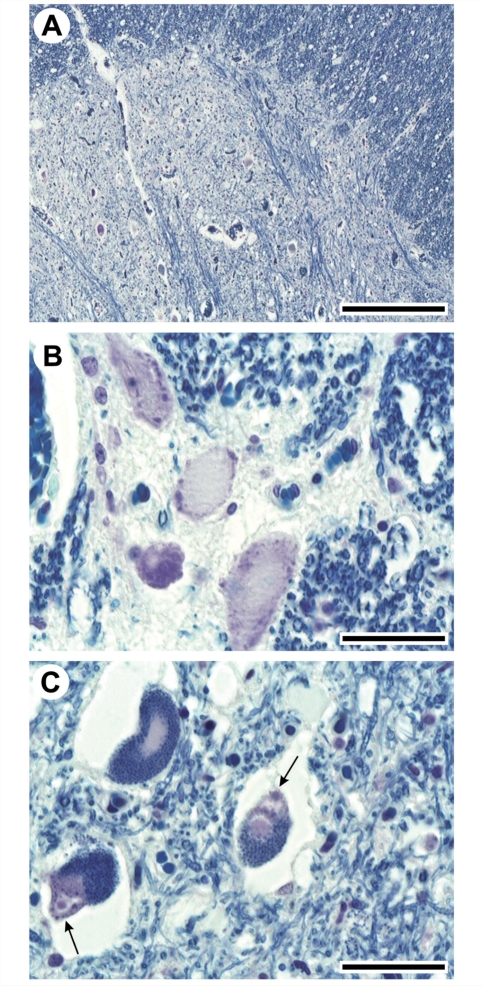
The mutation of the spatacsin gene is the single most common cause of autosomal recessive hereditary spastic paraplegia with thin corpus callosum. Common clinical, pathological and genetic features between amyotrophic lateral sclerosis and hereditary spastic paraplegia motivated us to investigate 25 families with autosomal recessive juvenile amyotrophic lateral sclerosis and long-term survival for mutations in the spatascin gene. The inclusion criterion was a diagnosis of clinically definite amyotrophic lateral sclerosis according to the revised El Escorial criteria. The exclusion criterion was a diagnosis of hereditary spastic paraplegia with thin corpus callosum in line with an established protocol. Additional pathological and genetic evaluations were also performed. Surprisingly, 12 sequence alterations in the spatacsin gene (one of which is novel, IVS30 + 1 G > A) were identified in 10 unrelated pedigrees with autosomal recessive juvenile amyotrophic lateral sclerosis and long-term survival. The countries of origin of these families were Italy, Brazil, Canada, Japan and Turkey. The variants seemed to be pathogenic since they co-segregated with the disease in all pedigrees, were absent in controls and were associated with amyotrophic lateral sclerosis neuropathology in one member of one of these families for whom central nervous system tissue was available. Our study indicates that mutations in the spatascin gene could cause a much wider spectrum of clinical features than previously recognized, including autosomal recessive juvenile amyotrophic lateral sclerosis.
Figures
Similar articles
-
ALS5/SPG11/KIAA1840 mutations cause autosomal recessive axonal Charcot-Marie-Tooth disease.Brain. 2016 Jan;139(Pt 1):73-85. doi: 10.1093/brain/awv320. Epub 2015 Nov 10. Brain. 2016. PMID: 26556829 Free PMC article.
-
Chinese families with autosomal recessive hereditary spastic paraplegia caused by mutations in SPG11.BMC Neurol. 2020 Jan 3;20(1):2. doi: 10.1186/s12883-019-1593-y. BMC Neurol. 2020. PMID: 31900114 Free PMC article.
-
Motor neuron degeneration in spastic paraplegia 11 mimics amyotrophic lateral sclerosis lesions.Brain. 2016 Jun;139(Pt 6):1723-34. doi: 10.1093/brain/aww061. Epub 2016 Mar 25. Brain. 2016. PMID: 27016404 Free PMC article.
-
[What is the role of the genetic survey in amyotrophic lateral sclerosis?].Rev Neurol (Paris). 2006 Jun;162 Spec No 2:4S91-4S95. Rev Neurol (Paris). 2006. PMID: 17128094 French.
-
Juvenile Amyotrophic Lateral Sclerosis: A Review.Genes (Basel). 2021 Nov 30;12(12):1935. doi: 10.3390/genes12121935. Genes (Basel). 2021. PMID: 34946884 Free PMC article. Review.
Cited by
-
Amyotrophic Lateral Sclerosis: Insights and New Prospects in Disease Pathophysiology, Biomarkers and Therapies.Pharmaceuticals (Basel). 2024 Oct 18;17(10):1391. doi: 10.3390/ph17101391. Pharmaceuticals (Basel). 2024. PMID: 39459030 Free PMC article. Review.
-
Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing.J Med Genet. 2013 Nov;50(11):776-83. doi: 10.1136/jmedgenet-2013-101795. Epub 2013 Jul 23. J Med Genet. 2013. PMID: 23881933 Free PMC article.
-
Genetic and phenotypic characterization of complex hereditary spastic paraplegia.Brain. 2016 Jul;139(Pt 7):1904-18. doi: 10.1093/brain/aww111. Epub 2016 May 23. Brain. 2016. PMID: 27217339 Free PMC article.
-
TDP-43 dysregulation and neuromuscular junction disruption in amyotrophic lateral sclerosis.Transl Neurodegener. 2022 Dec 27;11(1):56. doi: 10.1186/s40035-022-00331-z. Transl Neurodegener. 2022. PMID: 36575535 Free PMC article. Review.
-
Clinical genetics of amyotrophic lateral sclerosis: what do we really know?Nat Rev Neurol. 2011 Oct 11;7(11):603-15. doi: 10.1038/nrneurol.2011.150. Nat Rev Neurol. 2011. PMID: 21989245 Review.
References
-
- Ben Hamida M, Hentati F, Ben Hamida C. Hereditary motor system diseases (chronic juvenile amyotrophic lateral sclerosis). Conditions combining a bilateral pyramidal syndrome with limb and bulbar amyotrophy. Brain. 1990;113:347–63. - PubMed
-
- Boukhris A, Stevanin G, Feki I, Denis E, Elleuch N, Miladi MI, et al. Hereditary spastic paraplegia with mental impairment and thin corpus callosum in Tunisia: SPG11, SPG15, and further genetic heterogeneity. Arch Neurol. 2008;65:393–402. - PubMed
-
- Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–9. - PubMed
-
- Brugman F, Wokke JH, Scheffer H, Versteeg MH, Sistermans EA, van den Berg LH. Spastin mutations in sporadic adult-onset upper motor neuron syndromes. Ann Neurol. 2005;58:865–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
