

PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation
- PMID: 20107996
- PMCID: PMC2878662
- DOI: 10.1007/s00395-010-0084-5
PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation
Abstract
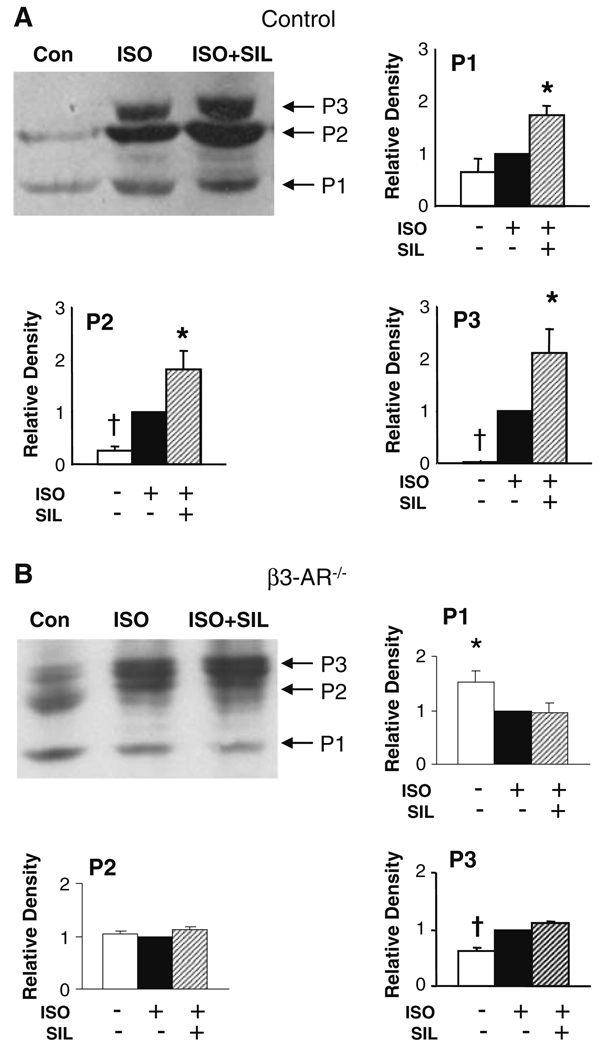
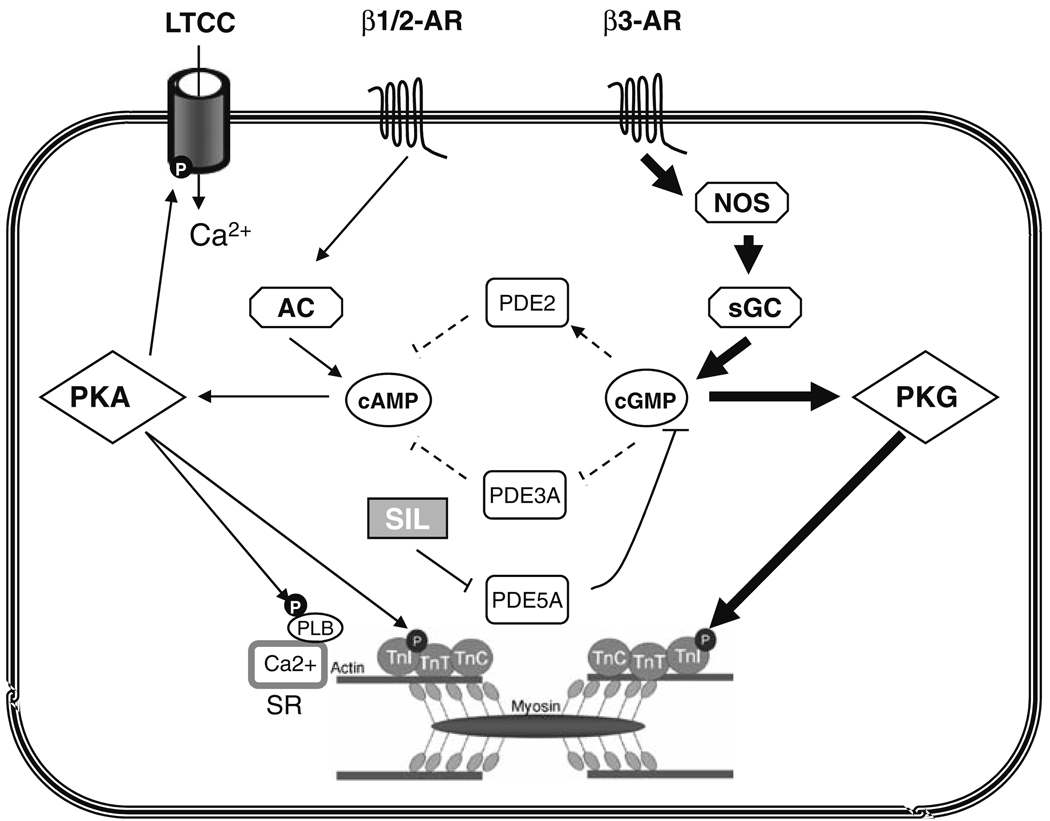
Phosphodiesterase type 5A (PDE5A) inhibitors acutely suppress beta-adrenergic receptor (beta-AR) stimulation in left ventricular myocytes and hearts. This modulation requires cyclic GMP synthesis via nitric oxide synthase (NOS)-NO stimulation, but upstream and downstream mechanisms remain un-defined. To determine this, adult cardiac myocytes from genetically engineered mice and controls were studied by video microscopy to assess sarcomere shortening (SS) and fura2-AM fluorescence to measure calcium transients (CaT). Enhanced SS from isoproterenol (ISO, 10 nM) was suppressed >or=50% by the PDE5A inhibitor sildenafil (SIL, 1 microM), without altering CaT. This regulation was unaltered despite co-inhibition of either the cGMP-stimulated cAMP-esterase PDE2 (Bay 60-7550), or cGMP-inhibited cAMP-esterase PDE3 (cilostamide). Thus, the SIL response could not be ascribed to cGMP interaction with alternative PDEs. However, genetic deletion (or pharmacologic blockade) of beta3-ARs, which couple to NOS signaling, fully prevented SIL modulation of ISO-stimulated SS. Importantly, both PDE5A protein expression and activity were similar in beta3-AR knockout (beta3-AR(-/-)) myocytes as in controls. Downstream, cGMP stimulates protein kinase G (PKG), and we found contractile modulation by SIL required PKG activation and enhanced TnI phosphorylation at S23, S24. Myocytes expressing the slow skeletal TnI isoform which lacks these sites displayed no modulation of ISO responses by SIL. Non-equilibrium isoelectric focusing gel electrophoresis showed SIL increased TnI phosphorylation above that from concomitant ISO in control but not beta3-AR(-/-) myocytes. These data support a cascade involving beta3-AR stimulation, and subsequent PKG-dependent TnI S23, S24 phosphorylation as primary factors underlying the capacity of acute PDE5A inhibition to blunt myocardial beta-adrenergic stimulation.
Conflict of interest statement
Figures
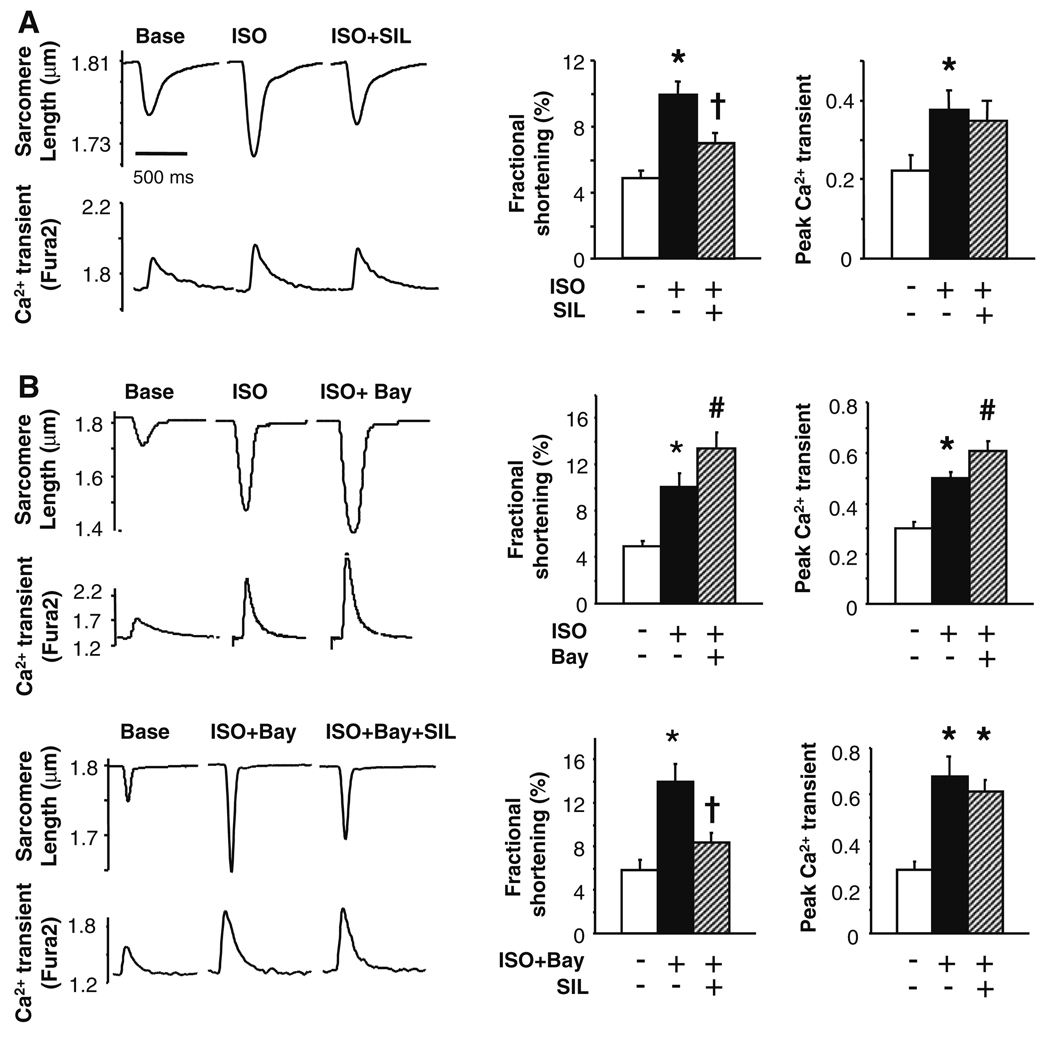
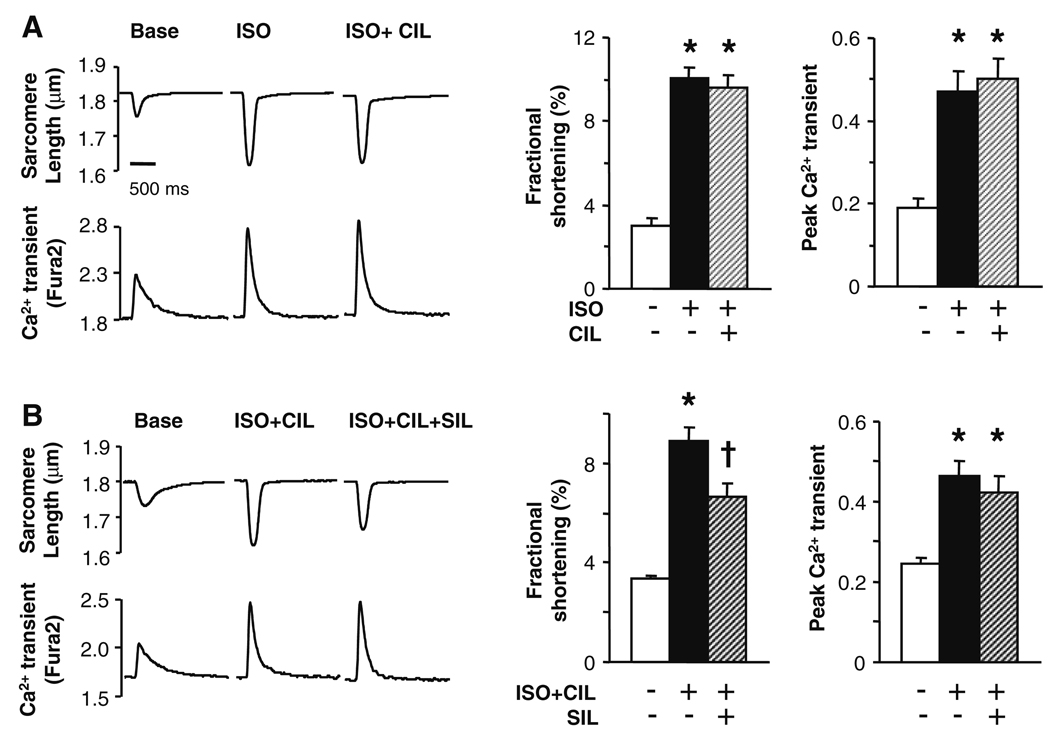
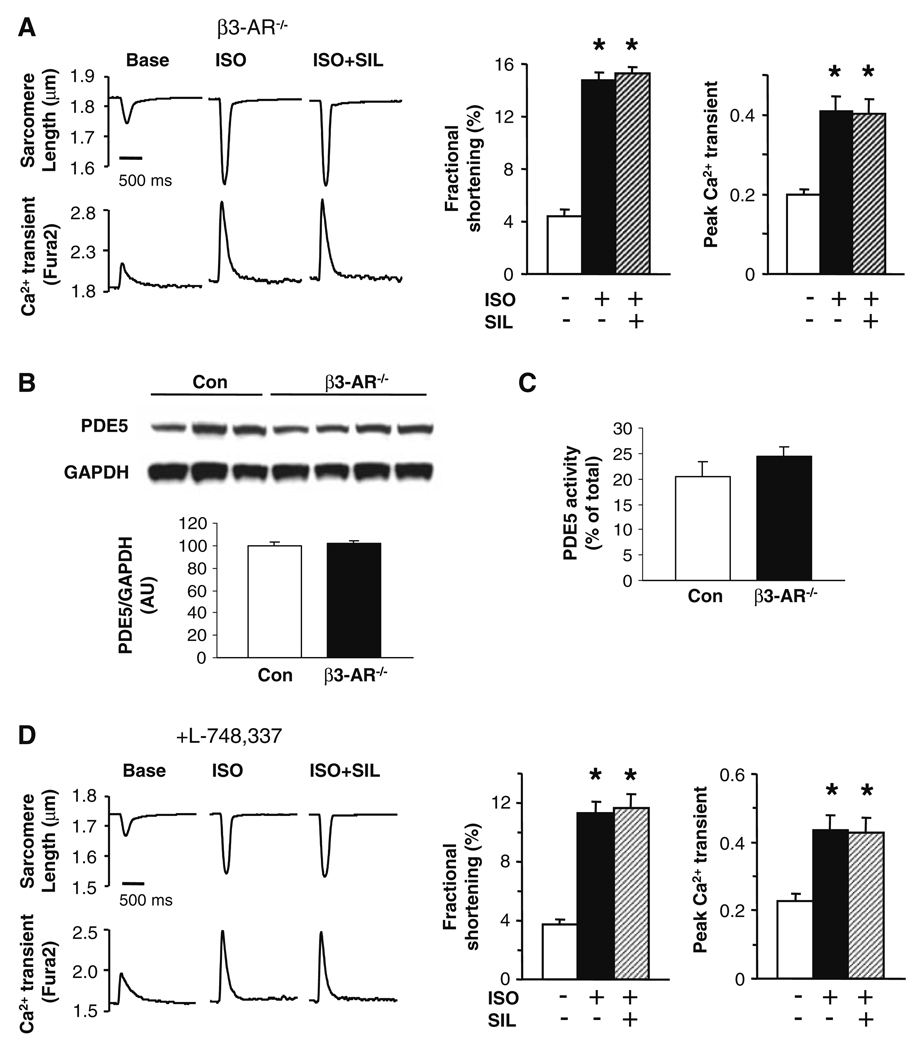
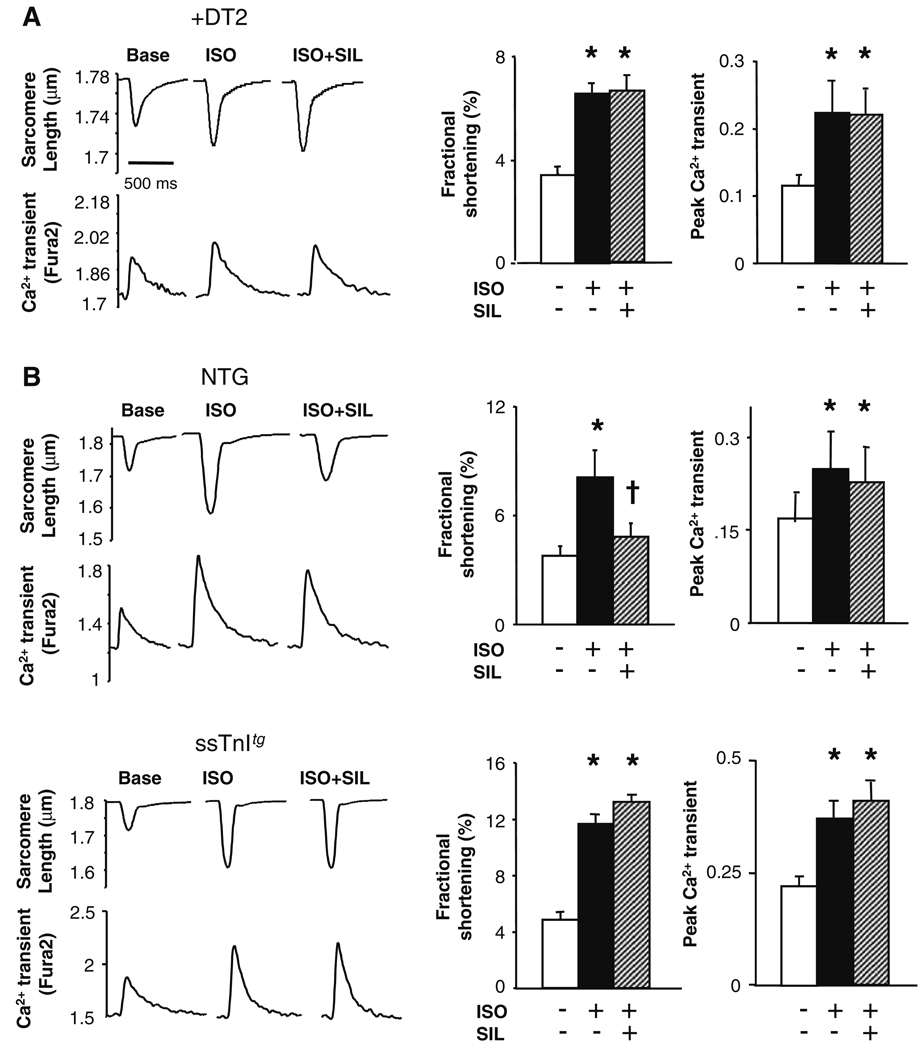
Similar articles
-
cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism.Circ Res. 2005 Jan 7;96(1):100-9. doi: 10.1161/01.RES.0000152262.22968.72. Epub 2004 Dec 2. Circ Res. 2005. PMID: 15576651
-
Compartmentalization of cardiac beta-adrenergic inotropy modulation by phosphodiesterase type 5.Circulation. 2007 Apr 24;115(16):2159-67. doi: 10.1161/CIRCULATIONAHA.106.643536. Epub 2007 Apr 9. Circulation. 2007. PMID: 17420342
-
Sustained soluble guanylate cyclase stimulation offsets nitric-oxide synthase inhibition to restore acute cardiac modulation by sildenafil.J Pharmacol Exp Ther. 2008 Aug;326(2):380-7. doi: 10.1124/jpet.108.137422. Epub 2008 May 2. J Pharmacol Exp Ther. 2008. PMID: 18456872
-
Regulation of phospholamban and troponin-I phosphorylation in the intact rat cardiomyocytes by adrenergic and cholinergic stimuli: roles of cyclic nucleotides, calcium, protein kinases and phosphatases and depolarization.Mol Cell Biochem. 1995 Aug-Sep;149-150:103-26. doi: 10.1007/BF01076569. Mol Cell Biochem. 1995. PMID: 8569720 Review.
-
Species- and tissue-dependent effects of NO and cyclic GMP on cardiac ion channels.Comp Biochem Physiol A Mol Integr Physiol. 2005 Oct;142(2):136-43. doi: 10.1016/j.cbpb.2005.04.012. Epub 2005 May 31. Comp Biochem Physiol A Mol Integr Physiol. 2005. PMID: 15927494 Review.
Cited by
-
Heart failure with preserved ejection fraction: emerging drug strategies.J Cardiovasc Pharmacol. 2013 Jul;62(1):13-21. doi: 10.1097/FJC.0b013e31829a4e61. J Cardiovasc Pharmacol. 2013. PMID: 23714774 Free PMC article. Review.
-
Cardiac Effects of Phosphodiesterase-5 Inhibitors: Efficacy and Safety.Cardiovasc Drugs Ther. 2023 Aug;37(4):793-806. doi: 10.1007/s10557-021-07275-y. Epub 2021 Oct 15. Cardiovasc Drugs Ther. 2023. PMID: 34652581 Free PMC article. Review.
-
Phosphodiesterase-5 Inhibitors as Therapeutics for Cardiovascular Diseases: A Brief Review.Iran J Public Health. 2023 May;52(5):870-879. doi: 10.18502/ijph.v52i5.12704. Iran J Public Health. 2023. PMID: 37484720 Free PMC article. Review.
-
Metabolic changes in hypertrophic cardiomyopathies: scientific update from the Working Group of Myocardial Function of the European Society of Cardiology.Cardiovasc Res. 2018 Aug 1;114(10):1273-1280. doi: 10.1093/cvr/cvy147. Cardiovasc Res. 2018. PMID: 29912308 Free PMC article. Review.
-
Phosphodiesterase in heart and vessels: from physiology to diseases.Physiol Rev. 2024 Apr 1;104(2):765-834. doi: 10.1152/physrev.00015.2023. Epub 2023 Nov 16. Physiol Rev. 2024. PMID: 37971403 Free PMC article. Review.
References
-
- Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005;112:2642–2649. - PubMed
-
- Bremer YA, Salloum F, Ockaili R, Chou E, Moskowitz WB, Kukreja RC. Sildenafil citrate (viagra) induces cardioprotective effects after ischemia/reperfusion injury in infant rabbits. Pediatr Res. 2005;57:22–27. - PubMed
-
- Brixius K, Bloch W, Ziskoven C, Bolck B, Napp A, Pott C, Steinritz D, Jiminez M, Addicks K, Giacobino JP, Schwinger RH. Beta3-adrenergic eNOS stimulation in left ventricular murine myocardium. Can J Physiol Pharmacol. 2006;84:1051–1060. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 HL062426/HL/NHLBI NIH HHS/United States
- T32 HL007227-32/HL/NHLBI NIH HHS/United States
- P01 HL059408/HL/NHLBI NIH HHS/United States
- P01 HL062426-100001/HL/NHLBI NIH HHS/United States
- R01 HL022231/HL/NHLBI NIH HHS/United States
- HL-095408/HL/NHLBI NIH HHS/United States
- T32 HL007227/HL/NHLBI NIH HHS/United States
- T32 HL-07227/HL/NHLBI NIH HHS/United States
- P01-HL-062426/HL/NHLBI NIH HHS/United States
- R01 HL089297-02/HL/NHLBI NIH HHS/United States
- HL-089297/HL/NHLBI NIH HHS/United States
- R01 HL022231-32/HL/NHLBI NIH HHS/United States
- R01 HL089297-02S1/HL/NHLBI NIH HHS/United States
- R01 HL089297/HL/NHLBI NIH HHS/United States
- R01 HL-022231/HL/NHLBI NIH HHS/United States
- P01 HL059408-049002/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
