

Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1
- PMID: 20038678
- PMCID: PMC2806285
- DOI: 10.1083/jcb.200906084
Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1
Abstract
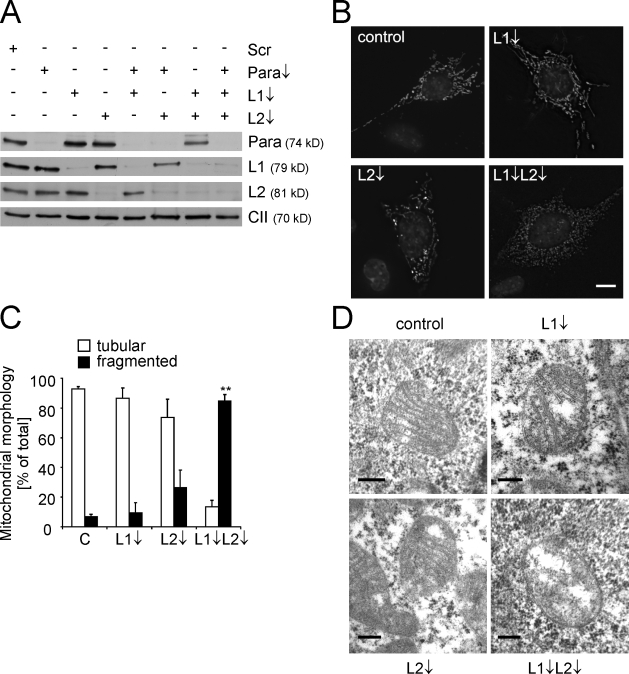
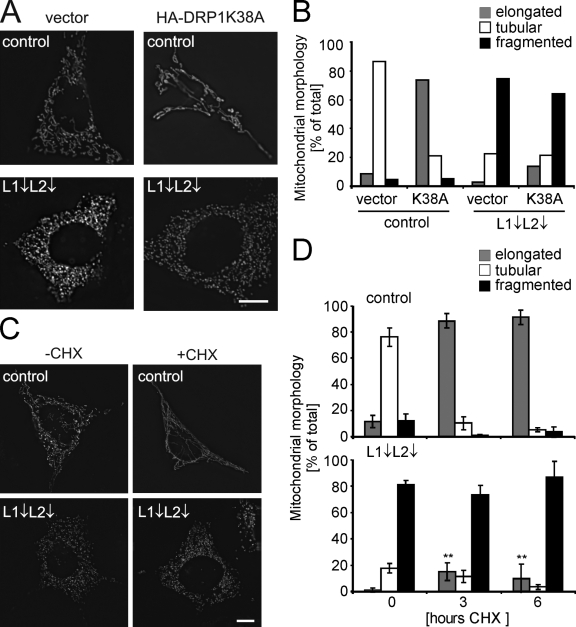
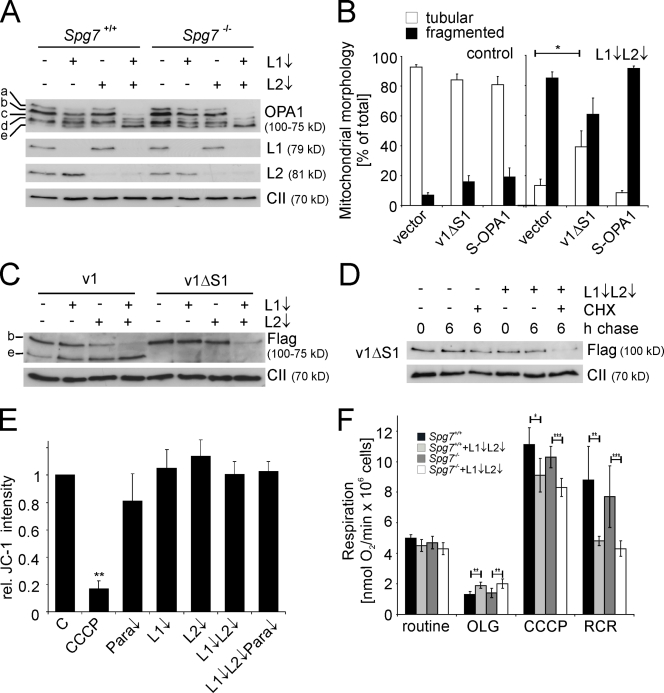
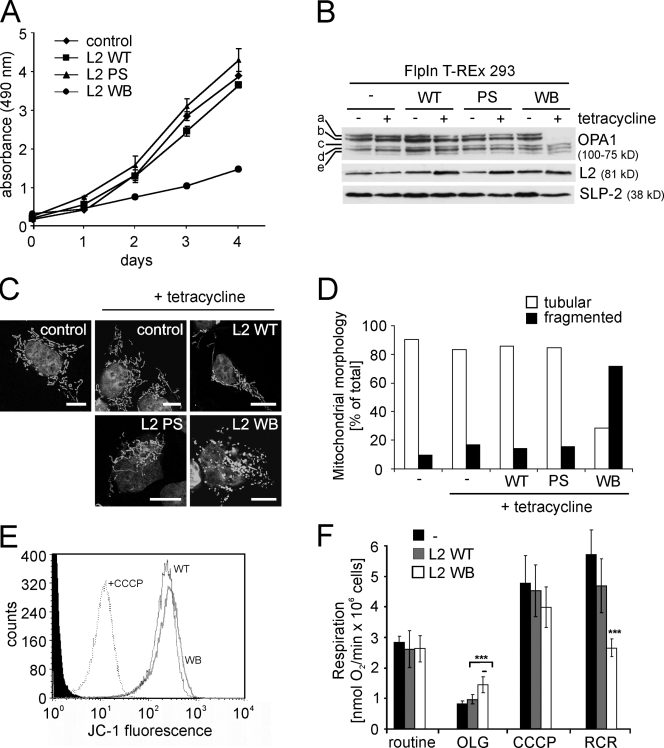
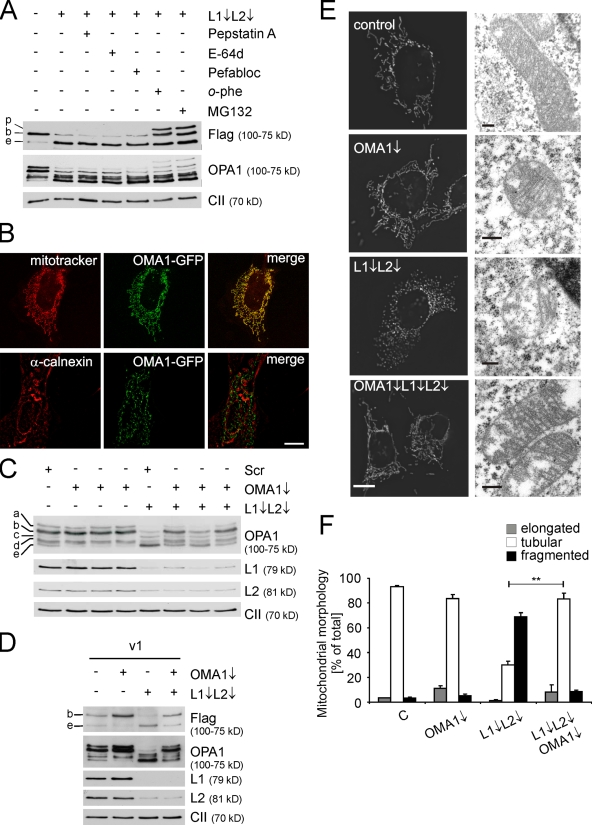
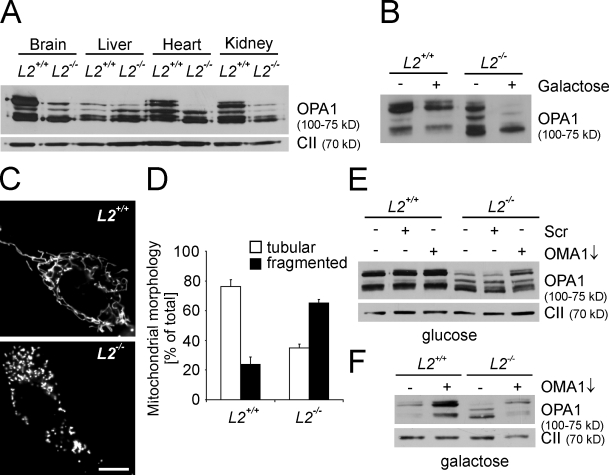
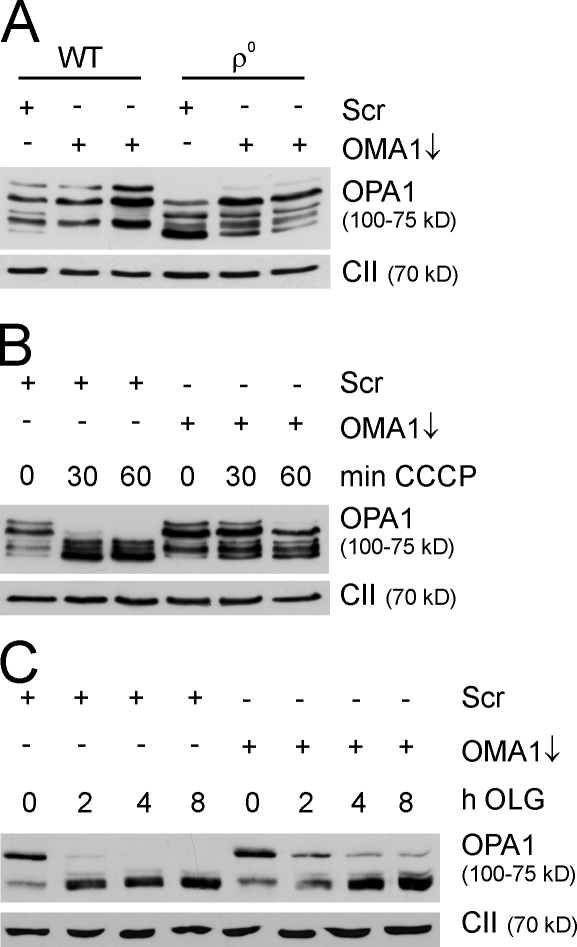
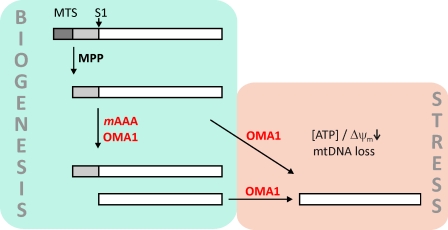
Mitochondrial fusion depends on the dynamin-like guanosine triphosphatase OPA1, whose activity is controlled by proteolytic cleavage. Dysfunction of mitochondria induces OPA1 processing and results in mitochondrial fragmentation, allowing the selective removal of damaged mitochondria. In this study, we demonstrate that two classes of metallopeptidases regulate OPA1 cleavage in the mitochondrial inner membrane: isoenzymes of the adenosine triphosphate (ATP)-dependent matrix AAA (ATPase associated with diverse cellular activities [m-AAA]) protease, variable assemblies of the conserved subunits paraplegin, AFG3L1 and -2, and the ATP-independent peptidase OMA1. Functionally redundant isoenzymes of the m-AAA protease ensure the balanced accumulation of long and short isoforms of OPA1 required for mitochondrial fusion. The loss of AFG3L2 in mouse tissues, down-regulation of AFG3L1 and -2 in mouse embryonic fibroblasts, or the expression of a dominant-negative AFG3L2 variant in human cells decreases the stability of long OPA1 isoforms and induces OPA1 processing by OMA1. Moreover, cleavage by OMA1 causes the accumulation of short OPA1 variants if mitochondrial DNA is depleted or mitochondrial activities are impaired. Our findings link distinct peptidases to constitutive and induced OPA1 processing and shed new light on the pathogenesis of neurodegenerative disorders associated with mutations in m-AAA protease subunits.
Figures
Similar articles
-
OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria.Mol Biol Cell. 2007 Sep;18(9):3582-90. doi: 10.1091/mbc.e07-02-0164. Epub 2007 Jul 5. Mol Biol Cell. 2007. PMID: 17615298 Free PMC article.
-
The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission.J Cell Biol. 2014 Mar 17;204(6):919-29. doi: 10.1083/jcb.201308006. Epub 2014 Mar 10. J Cell Biol. 2014. PMID: 24616225 Free PMC article.
-
Autocatalytic processing of m-AAA protease subunits in mitochondria.Mol Biol Cell. 2009 Oct;20(19):4216-24. doi: 10.1091/mbc.e09-03-0218. Epub 2009 Aug 5. Mol Biol Cell. 2009. PMID: 19656850 Free PMC article.
-
m-AAA proteases, mitochondrial calcium homeostasis and neurodegeneration.Cell Res. 2018 Mar;28(3):296-306. doi: 10.1038/cr.2018.17. Epub 2018 Feb 16. Cell Res. 2018. PMID: 29451229 Free PMC article. Review.
-
OPA1 processing in cell death and disease - the long and short of it.J Cell Sci. 2016 Jun 15;129(12):2297-306. doi: 10.1242/jcs.159186. Epub 2016 May 17. J Cell Sci. 2016. PMID: 27189080 Review.
Cited by
-
Impaired flickering of the permeability transition pore causes SPG7 spastic paraplegia.EBioMedicine. 2020 Nov;61:103050. doi: 10.1016/j.ebiom.2020.103050. Epub 2020 Oct 9. EBioMedicine. 2020. PMID: 33045469 Free PMC article.
-
N-terminal cleavage of the mitochondrial fusion GTPase OPA1 occurs via a caspase-independent mechanism in cerebellar granule neurons exposed to oxidative or nitrosative stress.Brain Res. 2013 Feb 4;1494:28-43. doi: 10.1016/j.brainres.2012.12.001. Epub 2012 Dec 7. Brain Res. 2013. PMID: 23220553 Free PMC article.
-
Defective mitophagy in Alzheimer's disease.Ageing Res Rev. 2020 Dec;64:101191. doi: 10.1016/j.arr.2020.101191. Epub 2020 Oct 3. Ageing Res Rev. 2020. PMID: 33022416 Free PMC article. Review.
-
Mitochondria: Insights into Crucial Features to Overcome Cancer Chemoresistance.Int J Mol Sci. 2021 Apr 30;22(9):4770. doi: 10.3390/ijms22094770. Int J Mol Sci. 2021. PMID: 33946271 Free PMC article. Review.
-
Pharmacological Progress of Mitophagy Regulation.Curr Neuropharmacol. 2023;21(5):1026-1041. doi: 10.2174/1570159X21666230314140528. Curr Neuropharmacol. 2023. PMID: 36918785 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
