

Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C
- PMID: 20037586
- PMCID: PMC2812627
- DOI: 10.1038/ng.512
Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C
Abstract
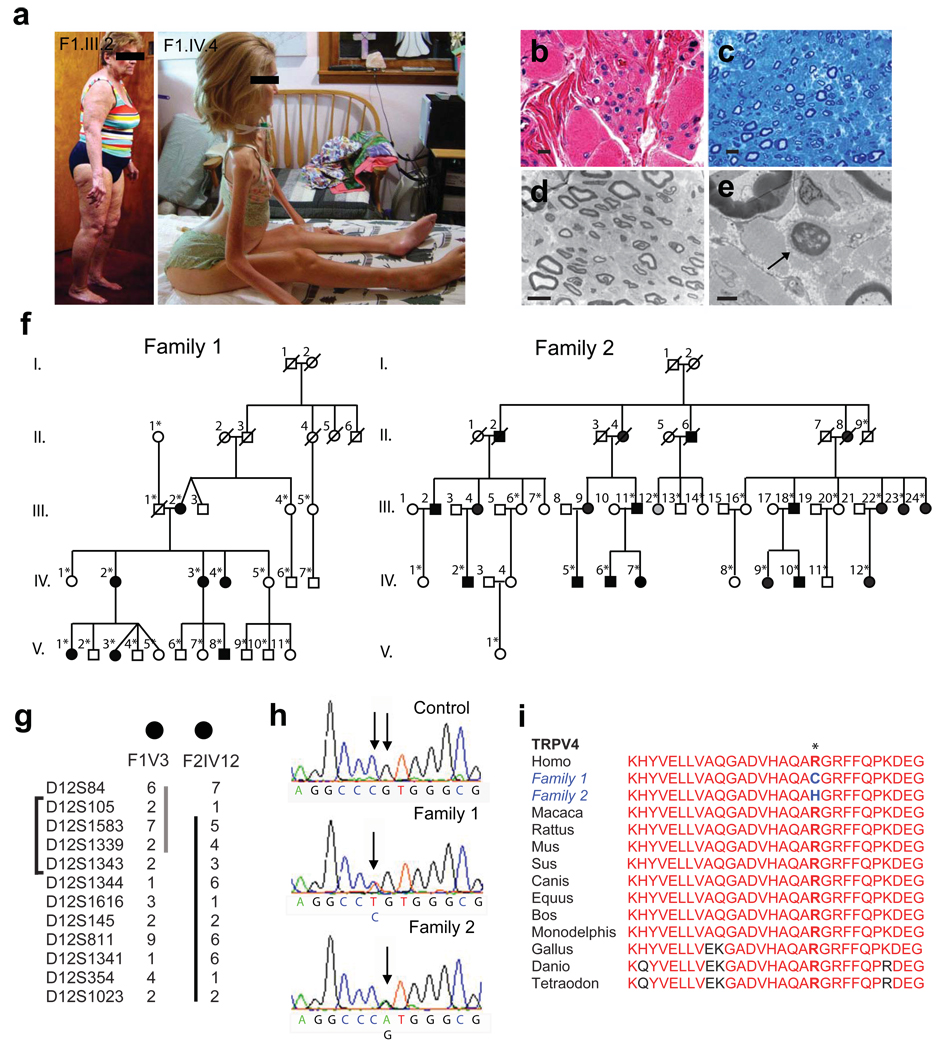
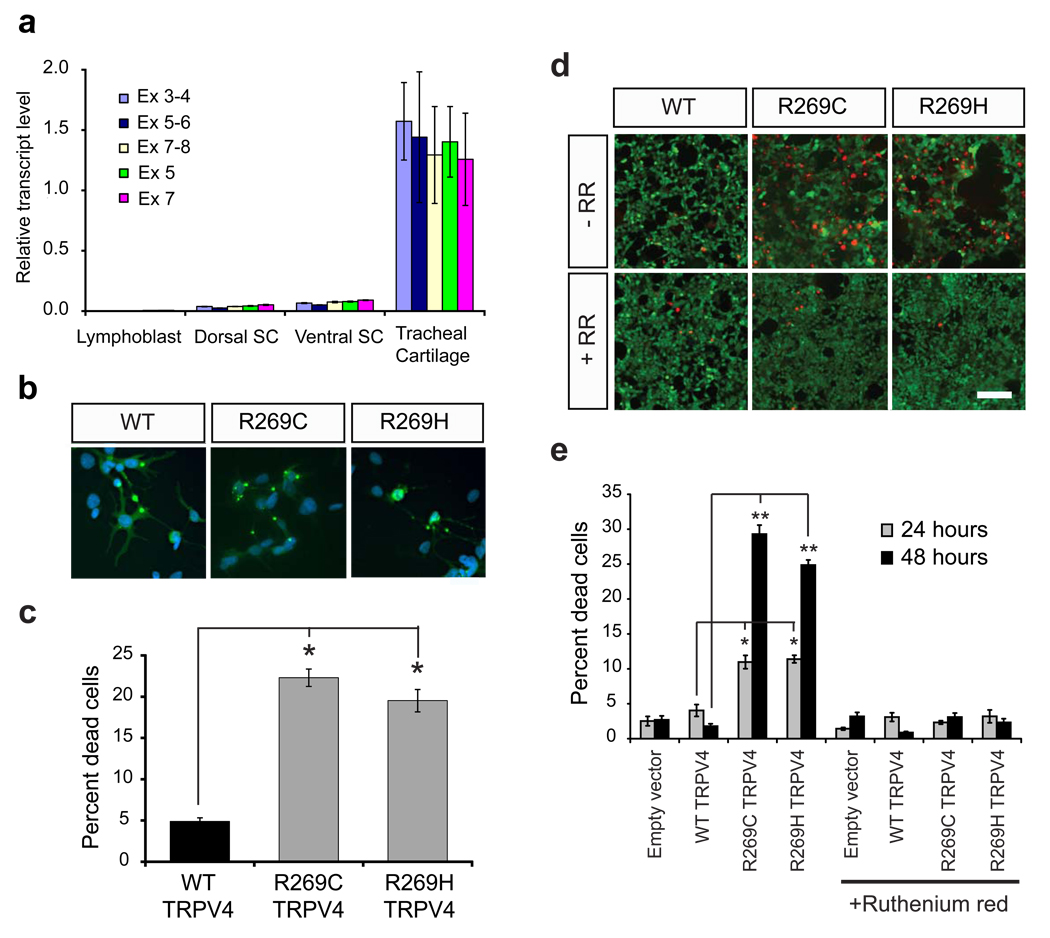
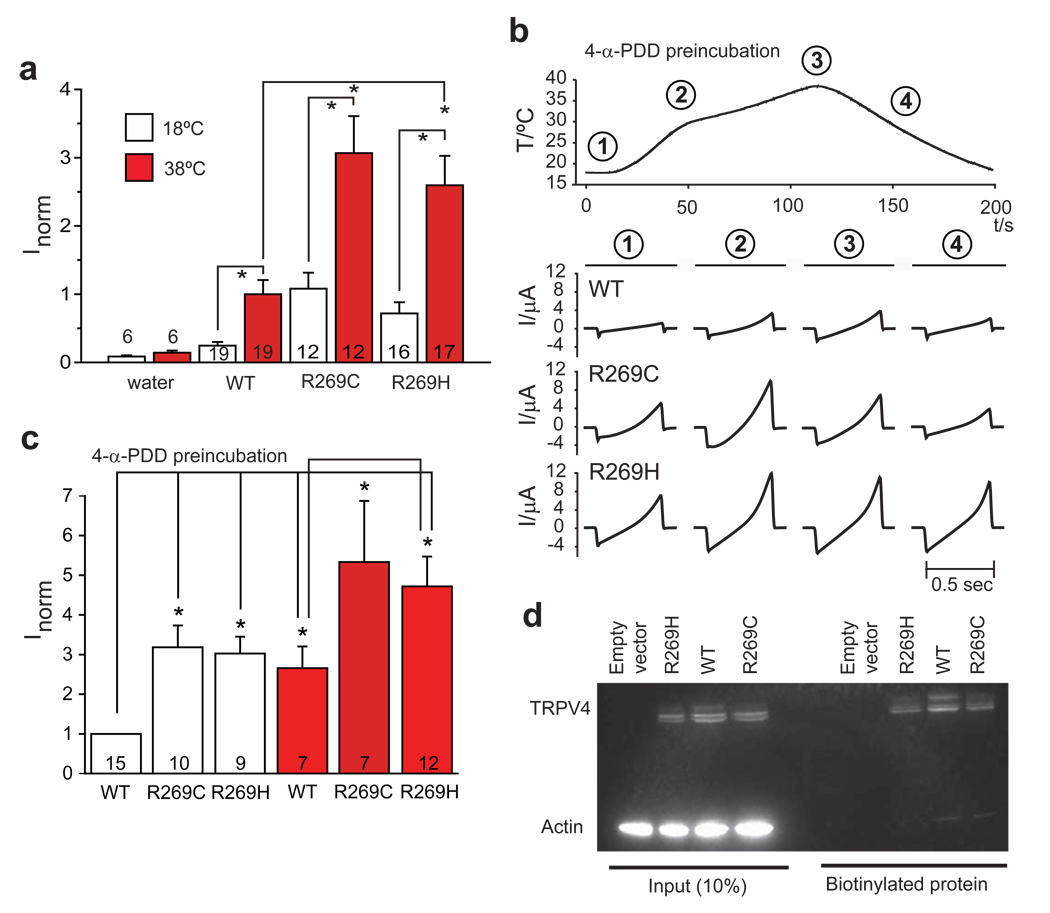
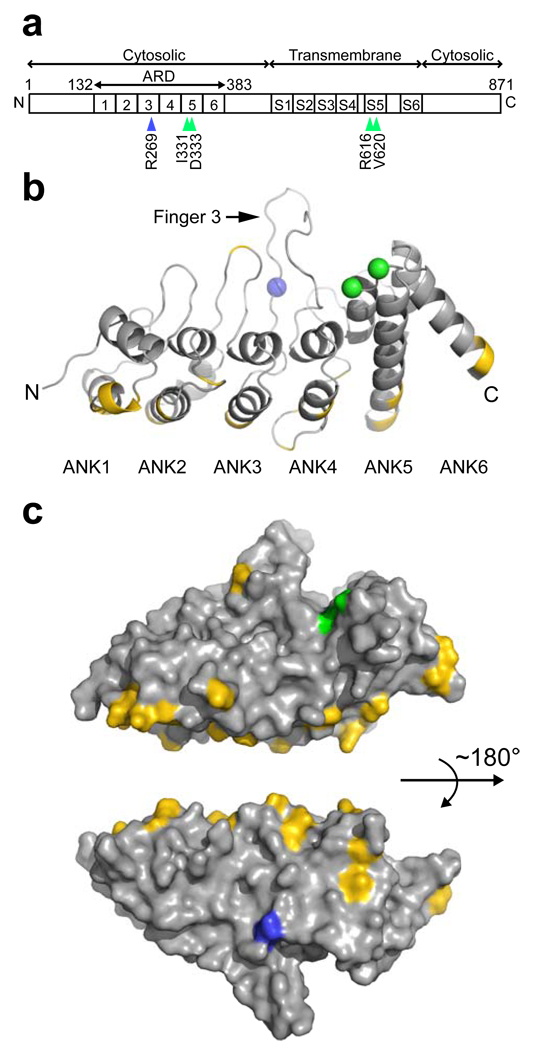
Charcot-Marie-Tooth disease type 2C (CMT2C) is an autosomal dominant neuropathy characterized by limb, diaphragm and laryngeal muscle weakness. Two unrelated families with CMT2C showed significant linkage to chromosome 12q24.11. We sequenced all genes in this region and identified two heterozygous missense mutations in the TRPV4 gene, C805T and G806A, resulting in the amino acid substitutions R269C and R269H. TRPV4 is a well-known member of the TRP superfamily of cation channels. In TRPV4-transfected cells, the CMT2C mutations caused marked cellular toxicity and increased constitutive and activated channel currents. Mutations in TRPV4 were previously associated with skeletal dysplasias. Our findings indicate that TRPV4 mutations can also cause a degenerative disorder of the peripheral nerves. The CMT2C-associated mutations lie in a distinct region of the TRPV4 ankyrin repeats, suggesting that this phenotypic variability may be due to differential effects on regulatory protein-protein interactions.
Figures
Comment in
-
Channelopathies converge on TRPV4.Nat Genet. 2010 Feb;42(2):98-100. doi: 10.1038/ng0210-98. Nat Genet. 2010. PMID: 20104247
-
Don't change that (calcium) channel: mutations in the same calcium channel gene can cause multiple distinct phenotypes.Clin Genet. 2010 Aug;78(2):134-6. doi: 10.1111/j.1399-0004.2010.01452_2.x. Clin Genet. 2010. PMID: 20662855 No abstract available.
Similar articles
-
Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4.Nat Genet. 2010 Feb;42(2):165-9. doi: 10.1038/ng.509. Epub 2009 Dec 27. Nat Genet. 2010. PMID: 20037587 Free PMC article.
-
Incidence and Clinical Features of TRPV4-Linked Axonal Neuropathies in a USA Cohort of Charcot-Marie-Tooth Disease Type 2.Neuromolecular Med. 2020 Mar;22(1):68-72. doi: 10.1007/s12017-019-08564-4. Epub 2019 Aug 29. Neuromolecular Med. 2020. PMID: 31468327
-
TRPV4 mutations and cytotoxic hypercalcemia in axonal Charcot-Marie-Tooth neuropathies.Neurology. 2011 Mar 8;76(10):887-94. doi: 10.1212/WNL.0b013e31820f2de3. Epub 2011 Feb 2. Neurology. 2011. PMID: 21288981 Free PMC article.
-
A TRPV4 mutation caused Charcot-Marie-Tooth disease type 2C with scapuloperoneal muscular atrophy overlap syndrome and scapuloperoneal spinal muscular atrophy in one family: a case report and literature review.BMC Neurol. 2023 Jun 30;23(1):250. doi: 10.1186/s12883-023-03260-0. BMC Neurol. 2023. PMID: 37391745 Free PMC article. Review.
-
Combined Phenotypes of Spondylometaphyseal Dysplasia-Kozlowski Type and Charcot-Marie-Tooth Disease Type 2C Secondary to a TRPV4 Pathogenic Variant.Mol Syndromol. 2019 May;10(3):154-160. doi: 10.1159/000495778. Epub 2018 Dec 21. Mol Syndromol. 2019. PMID: 31191204 Free PMC article. Review.
Cited by
-
The role of TRPV4 channels in ocular function and pathologies.Exp Eye Res. 2020 Dec;201:108257. doi: 10.1016/j.exer.2020.108257. Epub 2020 Sep 29. Exp Eye Res. 2020. PMID: 32979394 Free PMC article. Review.
-
TRPV4 is a regulator of adipose oxidative metabolism, inflammation, and energy homeostasis.Cell. 2012 Sep 28;151(1):96-110. doi: 10.1016/j.cell.2012.08.034. Cell. 2012. PMID: 23021218 Free PMC article.
-
Transient receptor potential channelopathies.Pflugers Arch. 2010 Jul;460(2):437-50. doi: 10.1007/s00424-010-0788-2. Epub 2010 Feb 4. Pflugers Arch. 2010. PMID: 20127491 Review.
-
Essential Tremor in a Charcot-Marie-Tooth Type 2C Kindred Does Not Segregate with the TRPV4 R269H Mutation.Case Rep Neurol. 2014 Jan 22;6(1):1-6. doi: 10.1159/000357665. eCollection 2014 Jan. Case Rep Neurol. 2014. PMID: 24575025 Free PMC article.
-
A Sporadic Case of Charcot-Marie-Tooth Disease Type 2 with Left Vocal Fold Palsy due to Mitofusin 2 Mutation.Intern Med. 2019 Jul 15;58(14):2091-2093. doi: 10.2169/internalmedicine.2318-18. Epub 2019 Apr 17. Intern Med. 2019. PMID: 30996168 Free PMC article.
References
-
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6:98–118. - PubMed
-
- Dyck PJ, et al. Hereditary motor and sensory neuropathy with diaphragm and vocal cord paresis. Ann Neurol. 1994;35:608–615. - PubMed
-
- Santoro L, et al. Charcot-Marie-Tooth disease type 2C: a distinct genetic entity. Clinical and molecular characterization of the first European family. Neuromuscul Disord. 2002;12:399–404. - PubMed
-
- McEntagart ME, et al. Confirmation of a hereditary motor and sensory neuropathy IIC locus at chromosome 12q23-q24. Ann Neurol. 2005;57:293–297. - PubMed
-
- Klein CJ, et al. The gene for HMSN2C maps to 12q23-24: a region of neuromuscular disorders. Neurology. 2003;60:1151–1156. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
