

Prion-like mechanisms in neurodegenerative diseases
- PMID: 20029438
- PMCID: PMC3648341
- DOI: 10.1038/nrn2786
Prion-like mechanisms in neurodegenerative diseases
Abstract
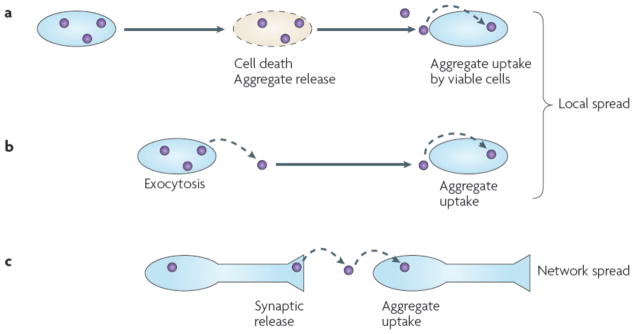
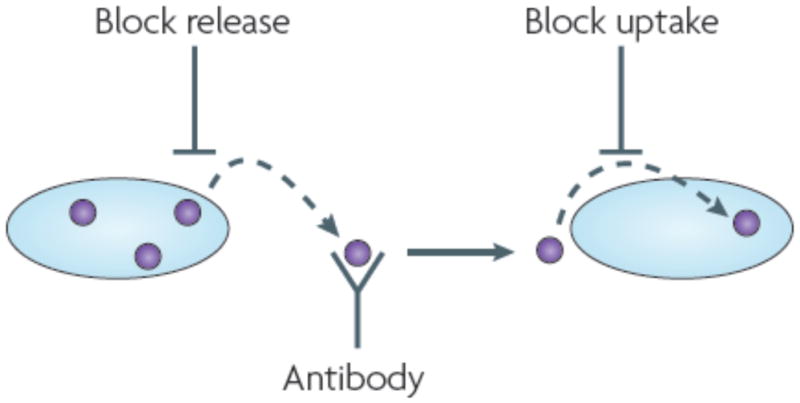
Many non-infectious neurodegenerative diseases are associated with the accumulation of fibrillar proteins. These diseases all exhibit features that are reminiscent of those of prionopathies, including phenotypic diversity and the propagation of pathology. Furthermore, emerging studies of amyloid-beta, alpha-synuclein and tau--proteins implicated in common neurodegenerative diseases--suggest that they share key biophysical and biochemical characteristics with prions. Propagation of protein misfolding in these diseases may therefore occur through mechanisms similar to those that underlie prion pathogenesis. If this hypothesis is verified in vivo, it will suggest new therapeutic strategies to block propagation of protein misfolding throughout the brain.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Prion-like propagation of protein aggregation and related therapeutic strategies.Neurotherapeutics. 2013 Jul;10(3):371-82. doi: 10.1007/s13311-013-0196-3. Neurotherapeutics. 2013. PMID: 23801258 Free PMC article. Review.
-
The expanding realm of prion phenomena in neurodegenerative disease.Prion. 2009 Apr-Jun;3(2):74-7. doi: 10.4161/pri.3.2.8754. Epub 2009 Apr 16. Prion. 2009. PMID: 19448400 Free PMC article. Review.
-
[The Propagation Hypothesis of Prion-like Protein Agregates in Neurodegenerative Diseases].Brain Nerve. 2019 Nov;71(11):1209-1214. doi: 10.11477/mf.1416201430. Brain Nerve. 2019. PMID: 31722306 Japanese.
-
Prion Diseases: A Unique Transmissible Agent or a Model for Neurodegenerative Diseases?Biomolecules. 2021 Feb 2;11(2):207. doi: 10.3390/biom11020207. Biomolecules. 2021. PMID: 33540845 Free PMC article. Review.
-
Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies.Curr Neurol Neurosci Rep. 2014 Nov;14(11):495. doi: 10.1007/s11910-014-0495-z. Curr Neurol Neurosci Rep. 2014. PMID: 25218483 Review.
Cited by
-
Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer.J Biol Chem. 2012 Aug 10;287(33):28152-62. doi: 10.1074/jbc.M112.340638. Epub 2012 Jun 19. J Biol Chem. 2012. PMID: 22715097 Free PMC article.
-
Molecular Mechanisms in the Pathogenesis of Alzheimer's disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation.Biomolecules. 2016 Apr 28;6(2):24. doi: 10.3390/biom6020024. Biomolecules. 2016. PMID: 27136595 Free PMC article. Review.
-
Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases.Int J Mol Sci. 2020 Sep 28;21(19):7174. doi: 10.3390/ijms21197174. Int J Mol Sci. 2020. PMID: 32998479 Free PMC article. Review.
-
Tau and Aβ42 in lavage fluid of pneumonia patients are associated with end-organ dysfunction: A prospective exploratory study.PLoS One. 2024 Feb 23;19(2):e0298816. doi: 10.1371/journal.pone.0298816. eCollection 2024. PLoS One. 2024. PMID: 38394060 Free PMC article.
-
Functional genomics approach for identification of molecular processes underlying neurodegenerative disorders in prion diseases.Curr Genomics. 2012 Aug;13(5):369-78. doi: 10.2174/138920212801619223. Curr Genomics. 2012. PMID: 23372423 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
