

Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links
- PMID: 20028736
- PMCID: PMC2820889
- DOI: 10.1128/MCB.00993-09
Differential roles for DNA polymerases eta, zeta, and REV1 in lesion bypass of intrastrand versus interstrand DNA cross-links
Abstract
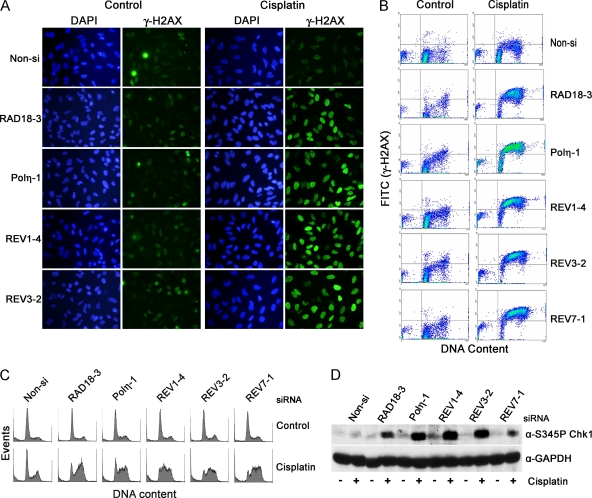
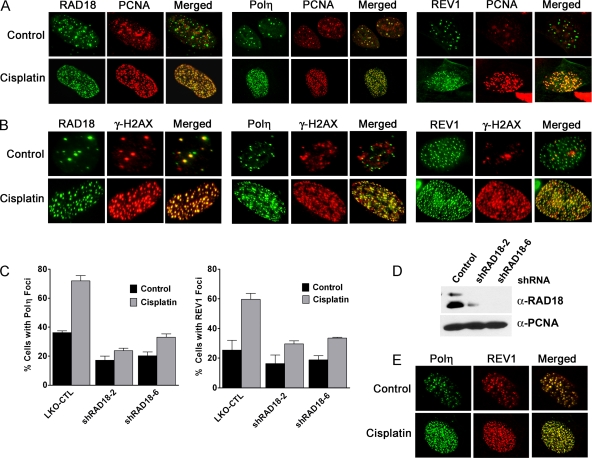
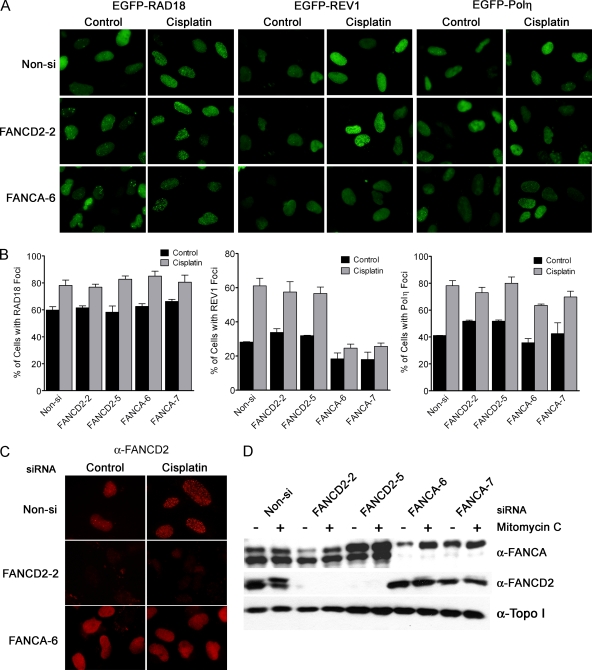
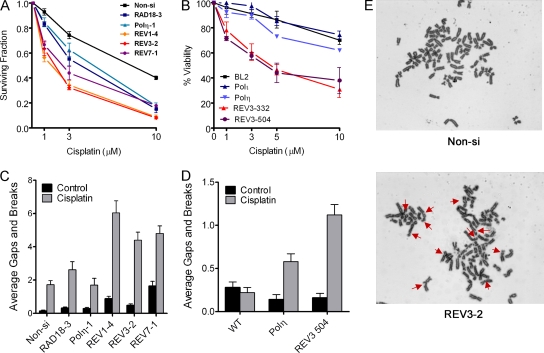
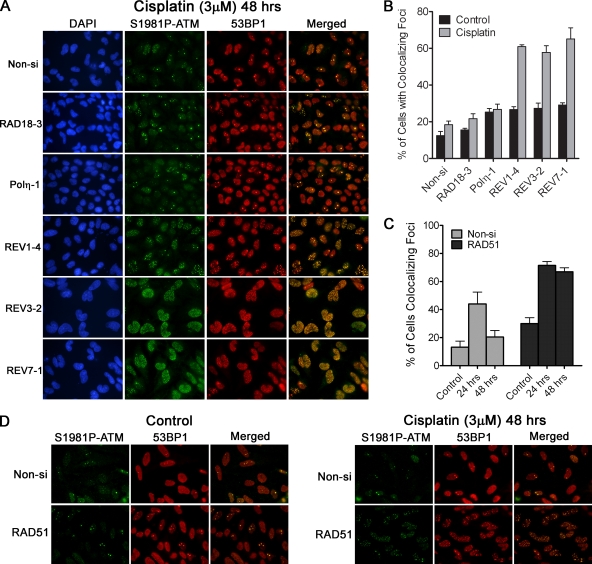
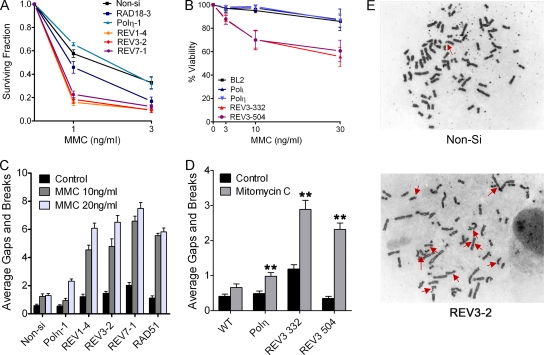
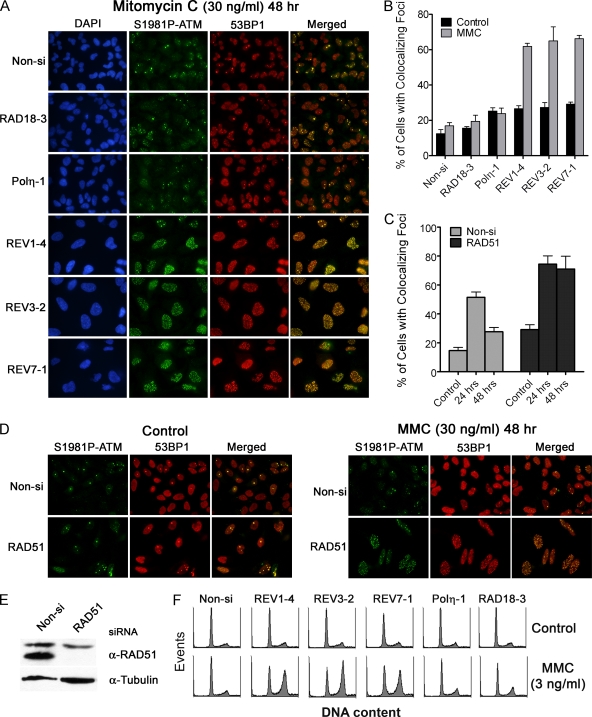
Translesion DNA synthesis (TLS) is a process whereby specialized DNA polymerases are recruited to bypass DNA lesions that would otherwise stall high-fidelity polymerases. We provide evidence that TLS across cisplatin intrastrand cross-links is performed by multiple translesion DNA polymerases. First, we determined that PCNA monoubiquitination by RAD18 is necessary for efficient bypass of cisplatin adducts by the TLS polymerases eta (Poleta), REV1, and zeta (Polzeta) based on the observations that depletion of these proteins individually leads to decreased cell survival, cell cycle arrest in S phase, and activation of the DNA damage response. Second, we showed that in addition to PCNA monoubiquitination by RAD18, the Fanconi anemia core complex is also important for recruitment of REV1 to stalled replication forks in cisplatin treated cells. Third, we present evidence that REV1 and Polzeta are uniquely associated with protection against cisplatin and mitomycin C-induced chromosomal aberrations, and both are necessary for the timely resolution of DNA double-strand breaks associated with repair of DNA interstrand cross-links. Together, our findings indicate that REV1 and Polzeta facilitate repair of interstrand cross-links independently of PCNA monoubiquitination and Poleta, whereas RAD18 plus Poleta, REV1, and Polzeta are all necessary for replicative bypass of cisplatin intrastrand DNA cross-links.
Figures

Similar articles
-
REV1 and polymerase ζ facilitate homologous recombination repair.Nucleic Acids Res. 2012 Jan;40(2):682-91. doi: 10.1093/nar/gkr769. Epub 2011 Sep 16. Nucleic Acids Res. 2012. PMID: 21926160 Free PMC article.
-
DNA polymerase ζ is a major determinant of resistance to platinum-based chemotherapeutic agents.Mol Pharmacol. 2012 Jun;81(6):778-87. doi: 10.1124/mol.111.076828. Epub 2012 Mar 2. Mol Pharmacol. 2012. PMID: 22387291 Free PMC article.
-
DNA-damage tolerance mediated by PCNA*Ub fusions in human cells is dependent on Rev1 but not Polη.Nucleic Acids Res. 2013 Aug;41(15):7356-69. doi: 10.1093/nar/gkt542. Epub 2013 Jun 12. Nucleic Acids Res. 2013. PMID: 23761444 Free PMC article.
-
Ubiquitin-dependent regulation of translesion polymerases.Biochem Soc Trans. 2010 Feb;38(Pt 1):110-5. doi: 10.1042/BST0380110. Biochem Soc Trans. 2010. PMID: 20074045 Review.
-
Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function.Annu Rev Biochem. 2005;74:317-53. doi: 10.1146/annurev.biochem.74.082803.133250. Annu Rev Biochem. 2005. PMID: 15952890 Review.
Cited by
-
Selective and cell-active inhibitors of the USP1/ UAF1 deubiquitinase complex reverse cisplatin resistance in non-small cell lung cancer cells.Chem Biol. 2011 Nov 23;18(11):1390-400. doi: 10.1016/j.chembiol.2011.08.014. Chem Biol. 2011. PMID: 22118673 Free PMC article.
-
Dual role for mammalian DNA polymerase ζ in maintaining genome stability and proliferative responses.Proc Natl Acad Sci U S A. 2013 Feb 19;110(8):E687-96. doi: 10.1073/pnas.1217425110. Epub 2013 Feb 5. Proc Natl Acad Sci U S A. 2013. PMID: 23386725 Free PMC article.
-
REV1 and polymerase ζ facilitate homologous recombination repair.Nucleic Acids Res. 2012 Jan;40(2):682-91. doi: 10.1093/nar/gkr769. Epub 2011 Sep 16. Nucleic Acids Res. 2012. PMID: 21926160 Free PMC article.
-
Chromatin Regulates Genome Targeting with Cisplatin.Angew Chem Int Ed Engl. 2017 Jun 1;56(23):6483-6487. doi: 10.1002/anie.201701144. Epub 2017 May 5. Angew Chem Int Ed Engl. 2017. PMID: 28474855 Free PMC article.
-
Co-inhibition of pol θ and HR genes efficiently synergize with cisplatin to suppress cisplatin-resistant lung cancer cells survival.Oncotarget. 2016 Oct 4;7(40):65157-65170. doi: 10.18632/oncotarget.11214. Oncotarget. 2016. PMID: 27533083 Free PMC article.
References
-
- Albertella, M. R., C. M. Green, A. R. Lehmann, and M. J. O'Connor. 2005. A role for polymerase η in the cellular tolerance to cisplatin-induced damage. Cancer Res. 65:9799-9806. - PubMed
-
- Alt, A., K. Lammens, C. Chiocchini, A. Lammens, J. C. Pieck, D. Kuch, K. P. Hopfner, and T. Carell. 2007. Bypass of DNA lesions generated during anticancer treatment with cisplatin by DNA polymerase η. Science 318:967-970. - PubMed
-
- Bakkenist, C. J., and M. B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499-506. - PubMed
-
- Bassett, E., N. M. King, M. F. Bryant, S. Hector, L. Pendyala, S. G. Chaney, and M. Cordeiro-Stone. 2004. The role of DNA polymerase η in translesion synthesis past Platinum-DNA adducts in human fibroblasts. Cancer Res. 64:6469-6475. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous