

FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling
- PMID: 20010692
- PMCID: PMC2829158
- DOI: 10.1038/emboj.2009.371
FANCM regulates DNA chain elongation and is stabilized by S-phase checkpoint signalling
Abstract
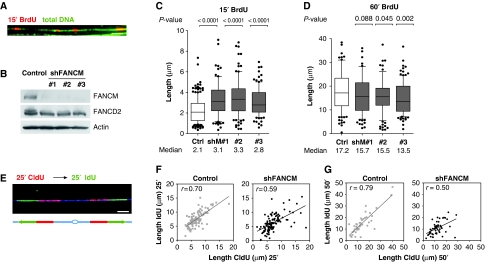
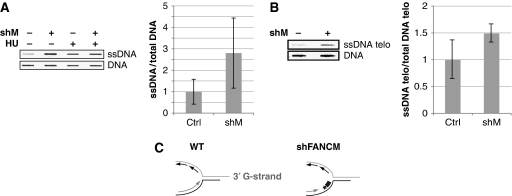
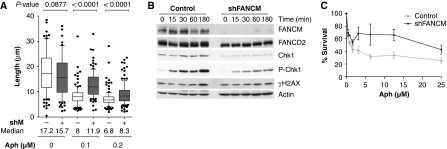
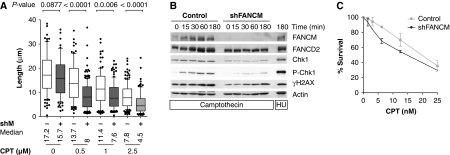
FANCM binds and remodels replication fork structures in vitro. We report that in vivo, FANCM controls DNA chain elongation in an ATPase-dependent manner. In the presence of replication inhibitors that do not damage DNA, FANCM counteracts fork movement, possibly by remodelling fork structures. Conversely, through damaged DNA, FANCM promotes replication and recovers stalled forks. Hence, the impact of FANCM on fork progression depends on the underlying hindrance. We further report that signalling through the checkpoint effector kinase Chk1 prevents FANCM from degradation by the proteasome after exposure to DNA damage. FANCM also acts in a feedback loop to stabilize Chk1. We propose that FANCM is a ringmaster in the response to replication stress by physically altering replication fork structures and by providing a tight link to S-phase checkpoint signalling.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Comment in
-
FANCM: fork pause, rewind and play.EMBO J. 2010 Feb 17;29(4):703-5. doi: 10.1038/emboj.2009.415. EMBO J. 2010. PMID: 20160754 Free PMC article.
Similar articles
-
ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions.Cancer Res. 2013 Jul 15;73(14):4300-10. doi: 10.1158/0008-5472.CAN-12-3976. Epub 2013 May 22. Cancer Res. 2013. PMID: 23698467 Free PMC article.
-
ATR activation and replication fork restart are defective in FANCM-deficient cells.EMBO J. 2010 Feb 17;29(4):806-18. doi: 10.1038/emboj.2009.385. Epub 2010 Jan 7. EMBO J. 2010. PMID: 20057355 Free PMC article.
-
FANCM regulates repair pathway choice at stalled replication forks.Mol Cell. 2021 Jun 3;81(11):2428-2444.e6. doi: 10.1016/j.molcel.2021.03.044. Epub 2021 Apr 20. Mol Cell. 2021. PMID: 33882298 Free PMC article.
-
FANCM branchpoint translocase: Master of traverse, reverse and adverse DNA repair.DNA Repair (Amst). 2024 Aug;140:103701. doi: 10.1016/j.dnarep.2024.103701. Epub 2024 Jun 4. DNA Repair (Amst). 2024. PMID: 38878565 Review.
-
The fork and the kinase: a DNA replication tale from a CHK1 perspective.Mutat Res Rev Mutat Res. 2015 Jan-Mar;763:168-80. doi: 10.1016/j.mrrev.2014.10.003. Epub 2014 Oct 22. Mutat Res Rev Mutat Res. 2015. PMID: 25795119 Free PMC article. Review.
Cited by
-
The Fml1-MHF complex suppresses inter-fork strand annealing in fission yeast.Elife. 2019 Dec 19;8:e49784. doi: 10.7554/eLife.49784. Elife. 2019. PMID: 31855181 Free PMC article.
-
The BLM dissolvasome in DNA replication and repair.Cell Mol Life Sci. 2013 Nov;70(21):4067-84. doi: 10.1007/s00018-013-1325-1. Epub 2013 Mar 31. Cell Mol Life Sci. 2013. PMID: 23543275 Free PMC article. Review.
-
FANCJ is essential to maintain microsatellite structure genome-wide during replication stress.Nucleic Acids Res. 2016 Aug 19;44(14):6803-16. doi: 10.1093/nar/gkw433. Epub 2016 May 13. Nucleic Acids Res. 2016. PMID: 27179029 Free PMC article.
-
Ubiquitin signalling in DNA replication and repair.Nat Rev Mol Cell Biol. 2010 Jul;11(7):479-89. doi: 10.1038/nrm2921. Epub 2010 Jun 16. Nat Rev Mol Cell Biol. 2010. PMID: 20551964 Review.
-
The Fanconi anemia pathway and ICL repair: implications for cancer therapy.Crit Rev Biochem Mol Biol. 2010 Oct;45(5):424-39. doi: 10.3109/10409238.2010.502166. Crit Rev Biochem Mol Biol. 2010. PMID: 20807115 Free PMC article. Review.
References
-
- Auerbach AD, Wolman SR (1976) Susceptibility of Fanconi's anaemia fibroblasts to chromosome damage by carcinogens. Nature 261: 494–496 - PubMed
-
- Azzalin CM, Lingner J (2006) The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr Biol 16: 433–439 - PubMed
-
- Bakker ST, van de Vrugt HJ, Rooimans MA, Oostra AB, Steltenpool J, Delzenne-Goette E, van der Wal A, van der Valk M, Joenje H, te Riele H, de Winter JP (2009) Fancm-deficient mice reveal unique features of Fanconi anemia complementation group M. Hum Mol Genet 18: 3484–3495 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
