

Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis
- PMID: 19940192
- PMCID: PMC2990681
- DOI: 10.1523/JNEUROSCI.3794-09.2009
Initiation and progression of axonopathy in experimental autoimmune encephalomyelitis
Abstract
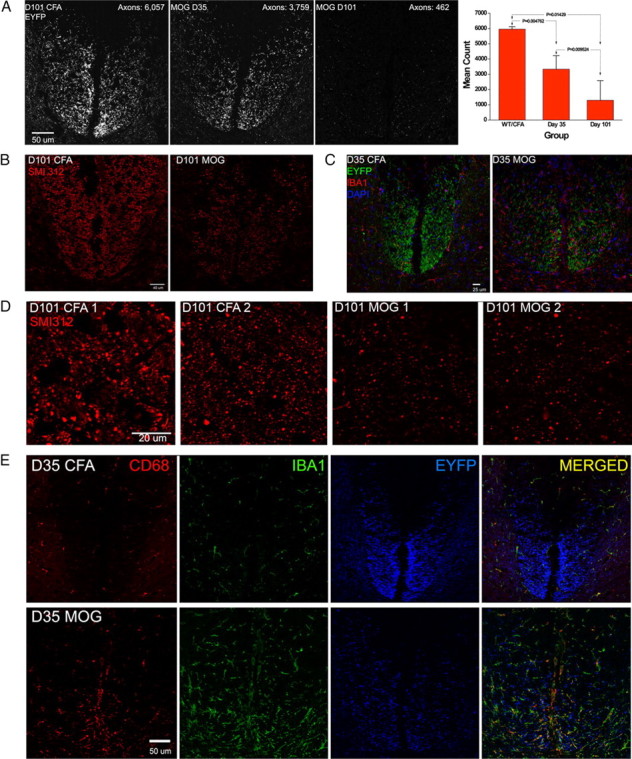
Axonal loss is the principal cause of chronic disability in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). In C57BL/6 mice with EAE induced by immunization with myelin oligodendrocyte glycoprotein peptide 35-55, the first evidences of axonal damage in spinal cord were in acute subpial and perivascular foci of infiltrating neutrophils and lymphocytes and included intra-axonal accumulations of the endovesicular Toll-like receptor TLR8, and the inflammasome protein NAcht leucine-rich repeat protein 1 (NALP1). Later in the course of this illness, focal inflammatory infiltrates disappeared from the spinal cord, but there was persistent activation of spinal cord innate immunity and progressive, bilaterally symmetric loss of small-diameter corticospinal tract axons. These results support the hypothesis that both contact-dependent and paracrine interactions of systemic inflammatory cells with axons and an innate immune-mediated neurodegenerative process contribute to axonal loss in this multiple sclerosis model.
Figures







Similar articles
-
Enhanced visualization of axonopathy in EAE using thy1-YFP transgenic mice.J Neurol Sci. 2007 Sep 15;260(1-2):23-32. doi: 10.1016/j.jns.2007.03.020. Epub 2007 May 9. J Neurol Sci. 2007. PMID: 17493638
-
Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation.Brain. 2012 Jun;135(Pt 6):1794-818. doi: 10.1093/brain/aws100. Epub 2012 Apr 28. Brain. 2012. PMID: 22544872 Free PMC article.
-
Time-Dependent Progression of Demyelination and Axonal Pathology in MP4-Induced Experimental Autoimmune Encephalomyelitis.PLoS One. 2015 Dec 11;10(12):e0144847. doi: 10.1371/journal.pone.0144847. eCollection 2015. PLoS One. 2015. PMID: 26658811 Free PMC article.
-
Validation of a novel biomarker for acute axonal injury in experimental autoimmune encephalomyelitis.J Neurosci Res. 2008 Dec;86(16):3548-55. doi: 10.1002/jnr.21803. J Neurosci Res. 2008. PMID: 18709652
-
Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease.J Neurol Sci. 2003 Feb 15;206(2):165-71. doi: 10.1016/s0022-510x(02)00069-2. J Neurol Sci. 2003. PMID: 12559505 Review.
Cited by
-
Hyaluronan anchored to activated CD44 on central nervous system vascular endothelial cells promotes lymphocyte extravasation in experimental autoimmune encephalomyelitis.J Biol Chem. 2012 Sep 28;287(40):33237-51. doi: 10.1074/jbc.M112.356287. Epub 2012 Aug 3. J Biol Chem. 2012. PMID: 22865853 Free PMC article.
-
The contribution of neutrophils to CNS autoimmunity.Clin Immunol. 2018 Apr;189:23-28. doi: 10.1016/j.clim.2016.06.017. Epub 2016 Jul 1. Clin Immunol. 2018. PMID: 27377536 Free PMC article. Review.
-
Antibodies to the RNA-binding protein hnRNP A1 contribute to neurodegeneration in a model of central nervous system autoimmune inflammatory disease.J Neuroinflammation. 2016 Jul 8;13(1):178. doi: 10.1186/s12974-016-0647-y. J Neuroinflammation. 2016. PMID: 27391474 Free PMC article.
-
Monitoring diffuse injury during disease progression in experimental autoimmune encephalomyelitis with on resonance variable delay multiple pulse (onVDMP) CEST MRI.Neuroimage. 2020 Jan 1;204:116245. doi: 10.1016/j.neuroimage.2019.116245. Epub 2019 Oct 9. Neuroimage. 2020. PMID: 31605825 Free PMC article.
-
Colony stimulating factors in the nervous system.Semin Immunol. 2021 Apr;54:101511. doi: 10.1016/j.smim.2021.101511. Epub 2021 Nov 4. Semin Immunol. 2021. PMID: 34743926 Free PMC article. Review.
References
-
- Aboul-Enein F, Weiser P, Höftberger R, Lassmann H, Bradl M. Transient axonal injury in the absence of demyelination: a correlate of clinical disease in acute experimental autoimmune encephalomyelitis. Acta Neuropathol. 2006;111:539–547. - PubMed
-
- Amadio S, Pluchino S, Brini E, Morana P, Guerriero R, Boneschi FM, Comi G, Zaratin P, Muzio V, del Carro U. Motor evoked potentials in a mouse model of chronic multiple sclerosis. Muscle Nerve. 2006;33:265–273. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous