

Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death
- PMID: 19850943
- PMCID: PMC2799045
- DOI: 10.1161/CIRCRESAHA.109.200378
Blockade of Hsp20 phosphorylation exacerbates cardiac ischemia/reperfusion injury by suppressed autophagy and increased cell death
Abstract
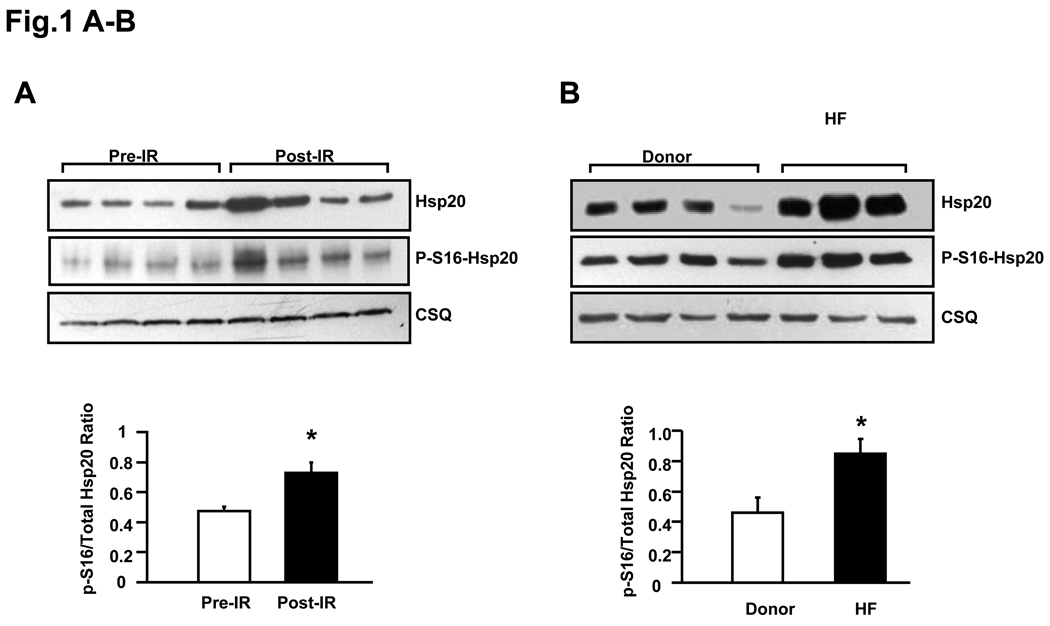
Rationale: The levels of a small heat shock protein (Hsp)20 and its phosphorylation are increased on ischemic insults, and overexpression of Hsp20 protects the heart against ischemia/reperfusion injury. However, the mechanism underlying cardioprotection of Hsp20 and especially the role of its phosphorylation in regulating ischemia/reperfusion-induced autophagy, apoptosis, and necrosis remain to be clarified.
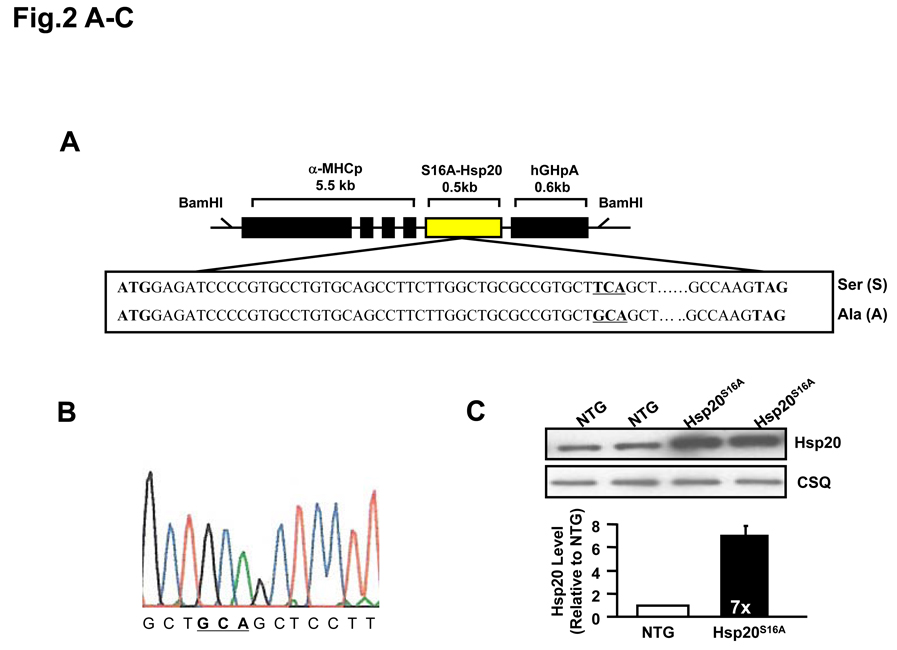
Objective: Herein, we generated a cardiac-specific overexpression model, carrying nonphosphorylatable Hsp20, where serine 16 was substituted with alanine (Hsp20(S16A)). By subjecting this model to ischemia/reperfusion, we addressed whether: (1) the cardioprotective effects of Hsp20 are associated with serine 16 phosphorylation; (2) blockade of Hsp20 phosphorylation influences the balance between autophagy and cell death; and (3) the aggregation pattern of Hsp20 is altered by its phosphorylation.
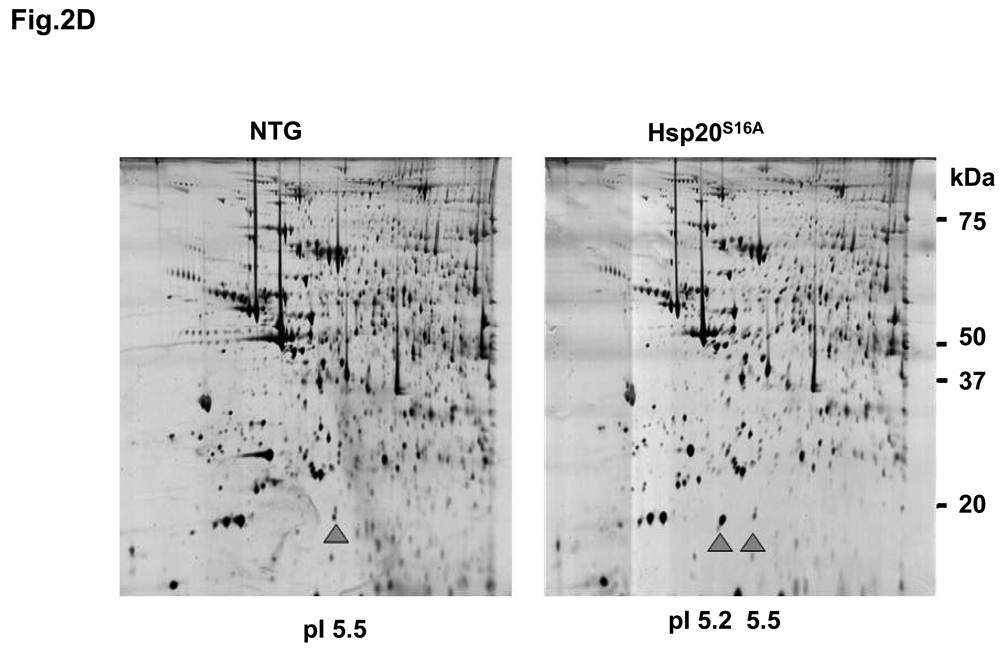
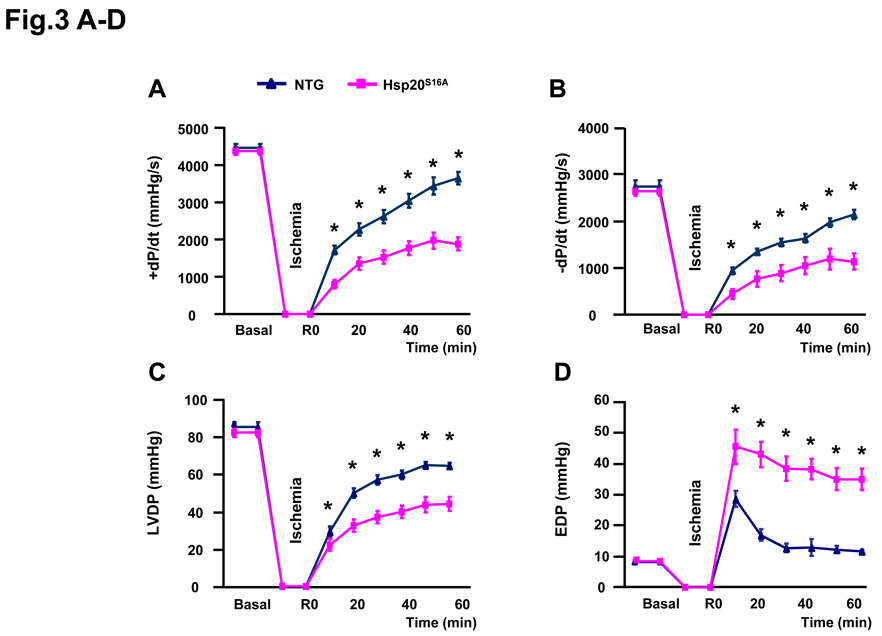
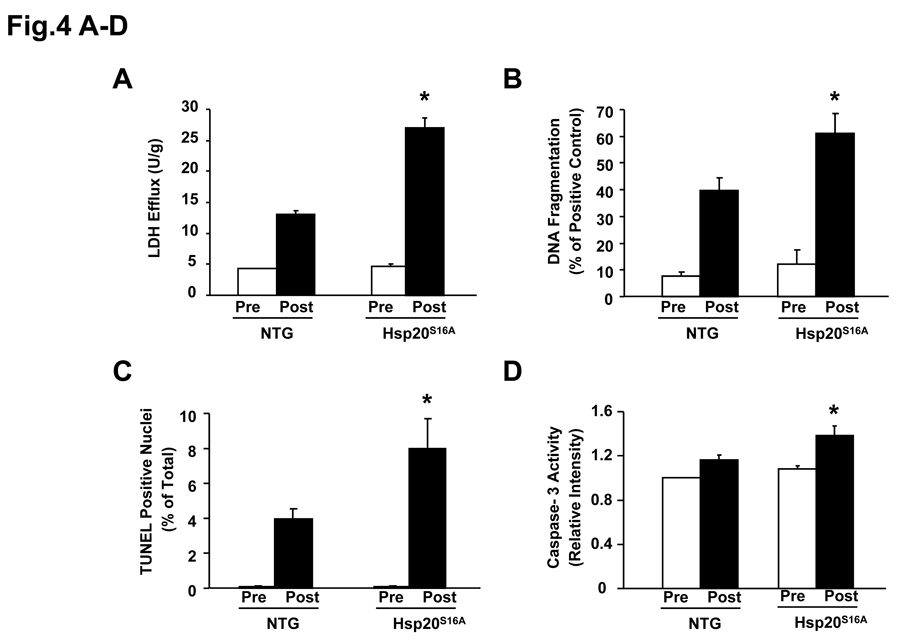
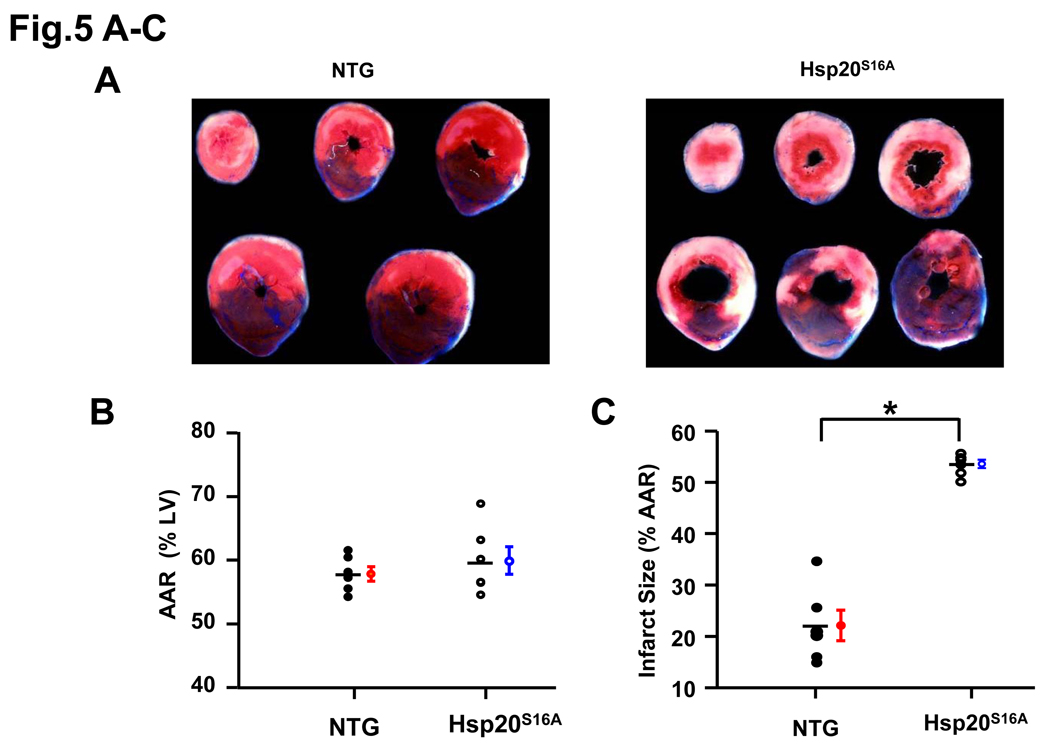
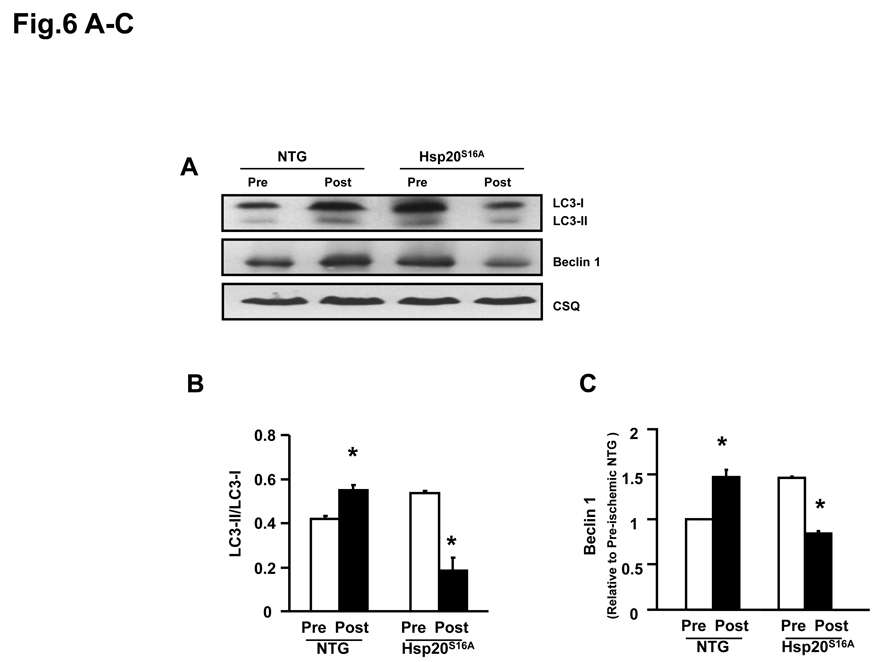
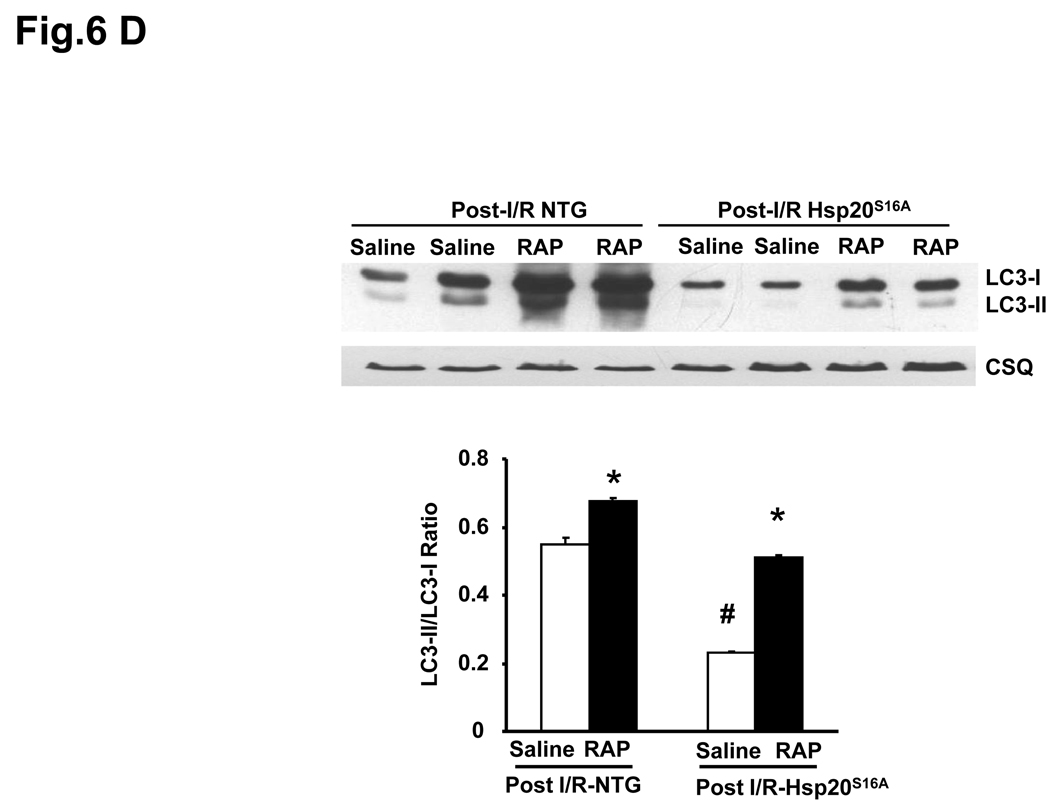
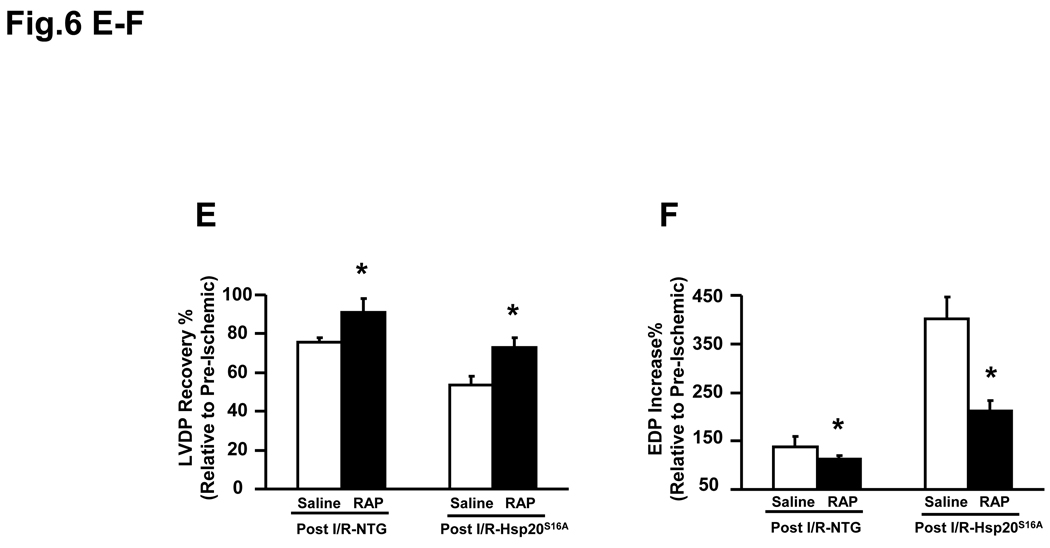
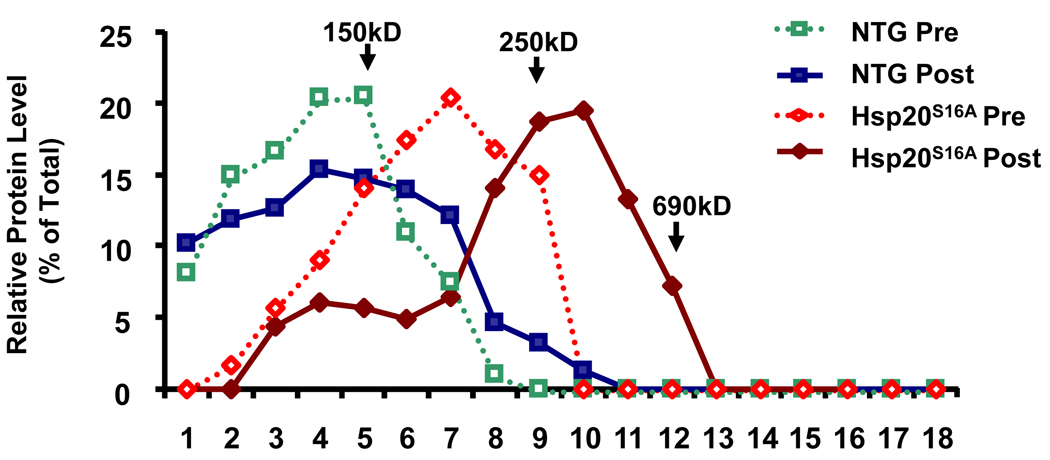
Methods and results: Our results demonstrated that Hsp20(S16A) hearts were more sensitive to ischemia/reperfusion injury, evidenced by lower recovery of contractile function and increased necrosis and apoptosis, compared with non-TG hearts. Interestingly, autophagy was activated in non-TG hearts but significantly inhibited in Hsp20(S16A) hearts following ischemia/reperfusion. Accordingly, pretreatment of Hsp20(S16A) hearts with rapamycin, an activator of autophagy, resulted in improvement of functional recovery, compared with saline-treated Hsp20(S16A) hearts. Furthermore, on ischemia/reperfusion, the oligomerization pattern of Hsp20 appeared to shift to higher aggregates in Hsp20(S16A) hearts.
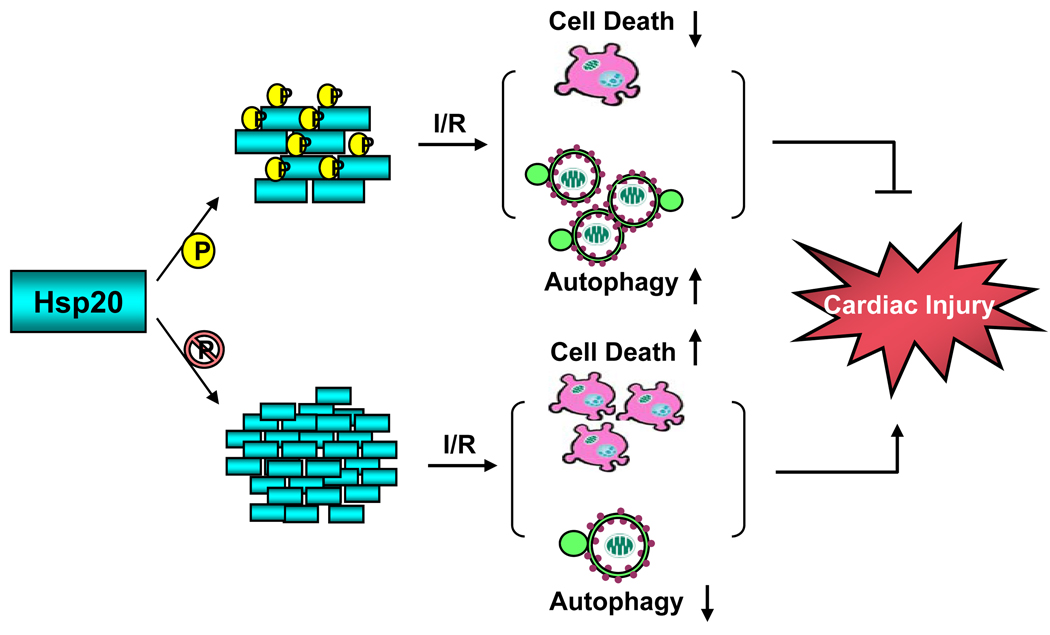
Conclusions: Collectively, these data indicate that blockade of Ser16-Hsp20 phosphorylation attenuates the cardioprotective effects of Hsp20 against ischemia/reperfusion injury, which may be attributable to suppressed autophagy and increased cell death. Therefore, phosphorylation of Hsp20 at serine 16 may represent a potential therapeutic target in ischemic heart disease.
Figures

Similar articles
-
Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury.Circulation. 2005 Apr 12;111(14):1792-9. doi: 10.1161/01.CIR.0000160851.41872.C6. Epub 2005 Apr 4. Circulation. 2005. PMID: 15809372
-
PKA phosphorylation of the small heat-shock protein Hsp20 enhances its cardioprotective effects.Biochem Soc Trans. 2012 Feb;40(1):210-4. doi: 10.1042/BST20110673. Biochem Soc Trans. 2012. PMID: 22260692 Free PMC article. Review.
-
Gene transfer of heat-shock protein 20 protects against ischemia/reperfusion injury in rat hearts.Acta Pharmacol Sin. 2005 Oct;26(10):1193-200. doi: 10.1111/j.1745-7254.2005.00139.x. Acta Pharmacol Sin. 2005. PMID: 16174435
-
Small heat shock protein 20 interacts with protein phosphatase-1 and enhances sarcoplasmic reticulum calcium cycling.Circ Res. 2011 Jun 10;108(12):1429-38. doi: 10.1161/CIRCRESAHA.110.237644. Epub 2011 Apr 14. Circ Res. 2011. PMID: 21493896 Free PMC article.
-
Hsp20 and its cardioprotection.Trends Cardiovasc Med. 2005 May;15(4):138-41. doi: 10.1016/j.tcm.2005.05.004. Trends Cardiovasc Med. 2005. PMID: 16099377 Review.
Cited by
-
Diabetes abolish cardioprotective effects of remote ischemic conditioning: evidences and possible mechanisms.J Physiol Biochem. 2019 Feb;75(1):19-28. doi: 10.1007/s13105-019-00664-w. Epub 2019 Feb 7. J Physiol Biochem. 2019. PMID: 30729392 Review.
-
Abnormal calcium cycling and cardiac arrhythmias associated with the human Ser96Ala genetic variant of histidine-rich calcium-binding protein.J Am Heart Assoc. 2013 Oct 14;2(5):e000460. doi: 10.1161/JAHA.113.000460. J Am Heart Assoc. 2013. PMID: 24125847 Free PMC article.
-
Circular RNA-circPan3 attenuates cardiac hypertrophy via miR-320-3p/HSP20 axis.Cell Mol Biol Lett. 2024 Jan 3;29(1):3. doi: 10.1186/s11658-023-00520-2. Cell Mol Biol Lett. 2024. PMID: 38172650 Free PMC article.
-
Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome.Circ Res. 2012 Jun 8;110(12):1646-60. doi: 10.1161/CIRCRESAHA.111.259754. Circ Res. 2012. PMID: 22679139 Free PMC article. Review.
-
Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde.Eur Heart J. 2011 Apr;32(8):1025-38. doi: 10.1093/eurheartj/ehq253. Epub 2010 Aug 12. Eur Heart J. 2011. PMID: 20705694 Free PMC article.
References
-
- Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res. 2001;51:637–646. - PubMed
-
- Benjamin IJ, McMillan D. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83:117–132. - PubMed
-
- Vander Heide RS. Increased expression of HSP27 protects canine myocytes from simulated ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2002;282:H935–H941. - PubMed
-
- Morrison LE, Whittaker RJ, Klepper RE, Wawrousek EF, Glembotski CC. Roles for alphaB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am J Physiol Heart Circ Physiol. 2004;286:H847–H855. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL-26057/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 HL087861-01/HL/NHLBI NIH HHS/United States
- R37 HL026057/HL/NHLBI NIH HHS/United States
- R01 HL087861/HL/NHLBI NIH HHS/United States
- R01 HL087861-03S1/HL/NHLBI NIH HHS/United States
- R01 HL087861-02/HL/NHLBI NIH HHS/United States
- HL-64018/HL/NHLBI NIH HHS/United States
- R01 HL026057/HL/NHLBI NIH HHS/United States
- HL-77101/HL/NHLBI NIH HHS/United States
- R01 HL064018/HL/NHLBI NIH HHS/United States
- R01 HL087861-03/HL/NHLBI NIH HHS/United States
- HL-087861/HL/NHLBI NIH HHS/United States
- P50 HL077101/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
