

Proteotoxic stress increases nuclear localization of ataxin-3
- PMID: 19843543
- PMCID: PMC2796889
- DOI: 10.1093/hmg/ddp482
Proteotoxic stress increases nuclear localization of ataxin-3
Abstract
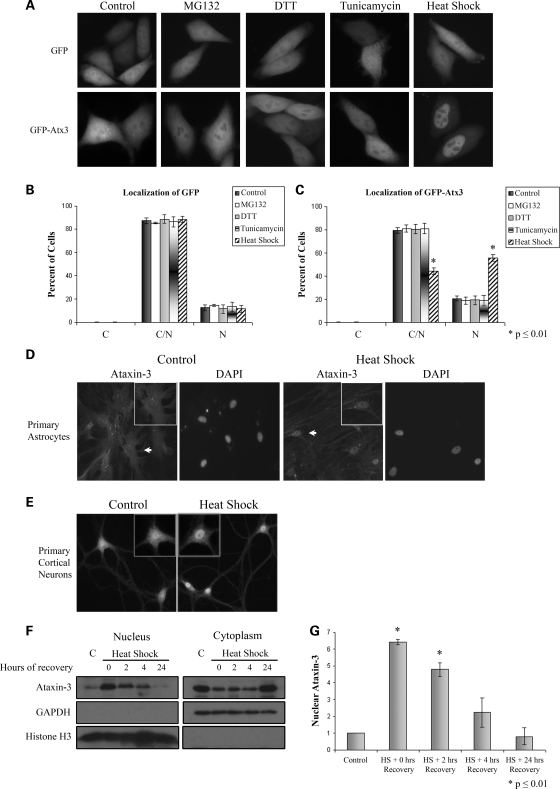
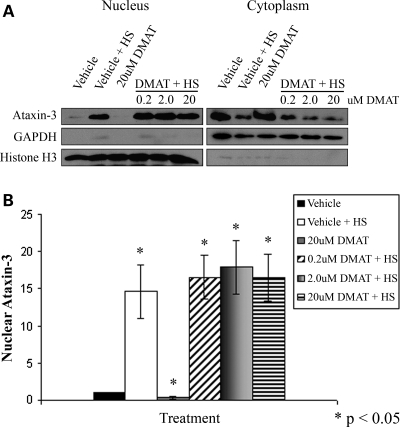
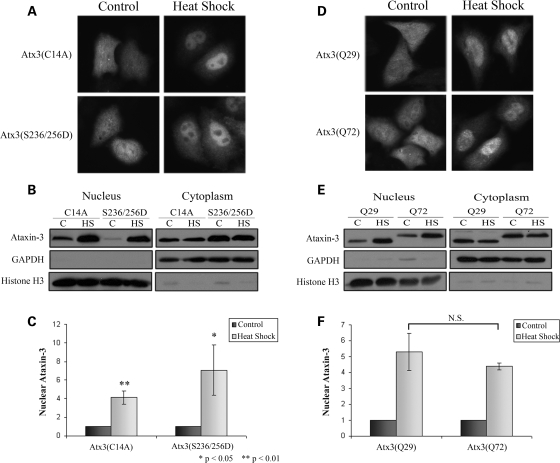
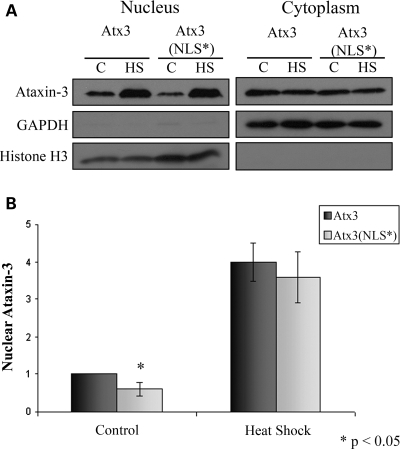
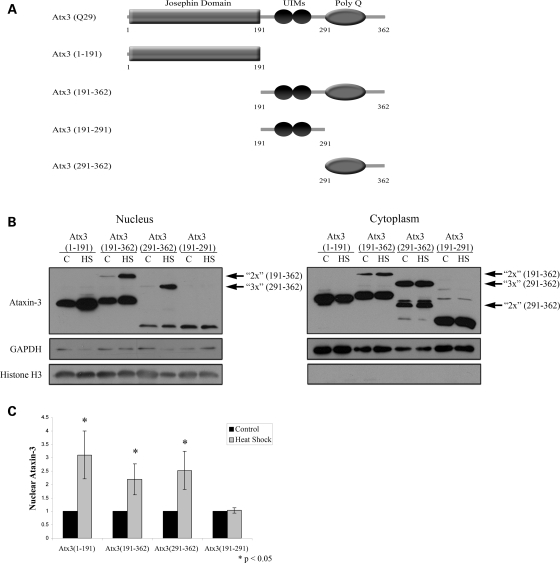
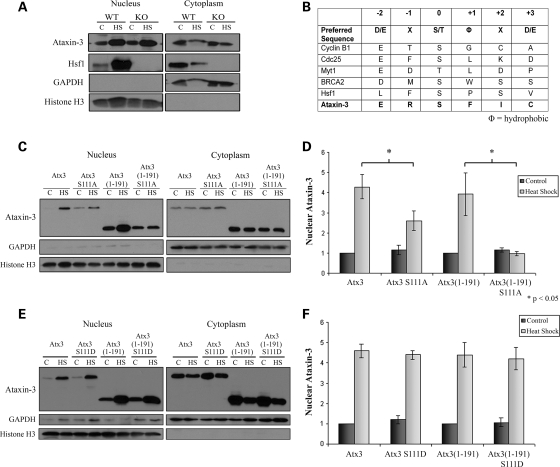
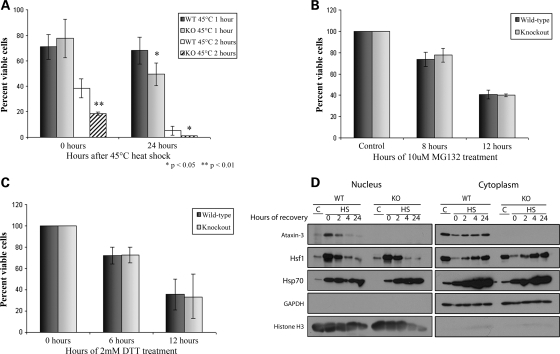
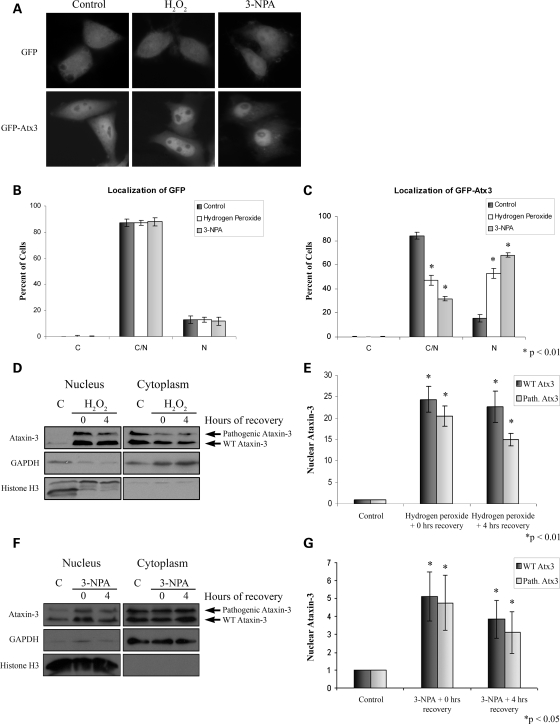
Spinocerebellar ataxia type 3 (SCA3)/Machado Joseph disease results from expansion of the polyglutamine domain in ataxin-3 (Atx3). Atx3 is a transcriptional co-repressor, as well as a deubiquitinating enzyme that appears to function in cellular pathways involved in protein homeostasis. In this study, we show that interactions of Atx3 with valosin-containing protein and hHR23B are dynamic and modulated by proteotoxic stresses. Heat shock, a general proteotoxic stress, also induced wild-type and pathogenic Atx3 to accumulate in the nucleus. Mapping studies showed that two regions of Atx3, the Josephin domain and the C-terminus, regulated heat shock-induced nuclear localization. Heat shock-induced nuclear localization of Atx3 was not affected by a casein kinase-2 inhibitor or by mutating a predicted nuclear localization signal. However, serine-111 of Atx3 was required for nuclear localization of the Josephin domain and regulated nuclear localization of full-length Atx3. Atx3 null cells were more sensitive to toxic effects of heat shock suggesting that Atx3 had a protective function in the cellular response to heat shock. Importantly, we found that oxidative stress also induced nuclear localization of Atx3; both wild-type and pathogenic Atx3 accumulated in the nucleus of SCA3 patient fibroblasts following oxidative stress. Heat shock and oxidative stress are the first processes identified that increase nuclear localization of Atx3. Observations in this study provide new and important insights for understanding SCA3 pathology as the nucleus is likely a key site for early pathogenesis.
Figures
Similar articles
-
Polyglutamine diseases: the special case of ataxin-3 and Machado-Joseph disease.Prog Neurobiol. 2011 Sep 15;95(1):26-48. doi: 10.1016/j.pneurobio.2011.06.007. Epub 2011 Jun 28. Prog Neurobiol. 2011. PMID: 21740957 Review.
-
CK2-dependent phosphorylation determines cellular localization and stability of ataxin-3.Hum Mol Genet. 2009 Sep 1;18(17):3334-43. doi: 10.1093/hmg/ddp274. Epub 2009 Jun 19. Hum Mol Genet. 2009. PMID: 19542537
-
Nucleocytoplasmic shuttling activity of ataxin-3.PLoS One. 2009 Jun 8;4(6):e5834. doi: 10.1371/journal.pone.0005834. PLoS One. 2009. PMID: 19503814 Free PMC article.
-
Flanking domain stability modulates the aggregation kinetics of a polyglutamine disease protein.Protein Sci. 2011 Oct;20(10):1675-81. doi: 10.1002/pro.698. Epub 2011 Aug 18. Protein Sci. 2011. PMID: 21780213 Free PMC article.
-
[Recent advances in molecular genetics of spinocerebellar ataxia type 3/Machado-Joseph disease].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2008 Dec;25(6):660-2. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2008. PMID: 19065526 Review. Chinese.
Cited by
-
Lentiviral vector-mediated overexpression of mutant ataxin-7 recapitulates SCA7 pathology and promotes accumulation of the FUS/TLS and MBNL1 RNA-binding proteins.Mol Neurodegener. 2016 Jul 28;11(1):58. doi: 10.1186/s13024-016-0123-2. Mol Neurodegener. 2016. PMID: 27465358 Free PMC article.
-
Degron capability of the hydrophobic C-terminus of the polyglutamine disease protein, ataxin-3.J Neurosci Res. 2020 Oct;98(10):2096-2108. doi: 10.1002/jnr.24684. Epub 2020 Jul 9. J Neurosci Res. 2020. PMID: 32643791 Free PMC article.
-
The de-ubiquitinating enzyme ataxin-3 does not modulate disease progression in a knock-in mouse model of Huntington disease.J Huntingtons Dis. 2013;2(2):201-15. doi: 10.3233/JHD-130058. J Huntingtons Dis. 2013. PMID: 24683430 Free PMC article.
-
Machado-Joseph Deubiquitinases: From Cellular Functions to Potential Therapy Targets.Front Pharmacol. 2020 Aug 26;11:1311. doi: 10.3389/fphar.2020.01311. eCollection 2020. Front Pharmacol. 2020. PMID: 32982735 Free PMC article. Review.
-
Anti-Excitotoxic Effects of N-Butylidenephthalide Revealed by Chemically Insulted Purkinje Progenitor Cells Derived from SCA3 iPSCs.Int J Mol Sci. 2022 Jan 26;23(3):1391. doi: 10.3390/ijms23031391. Int J Mol Sci. 2022. PMID: 35163312 Free PMC article.
References
-
- Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. - PubMed
-
- Paulson H.L., Perez M.K., Trottier Y., Trojanowski J.Q., Subramony S.H., Das S.S., Vig P., Mandel J.L., Fischbeck K.H., Pittman R.N. Intranuclear inclusions of expanded polyglutamine protein in spinocerebellar ataxia type 3. Neuron. 1997;19:333–344. - PubMed
-
- Davies S.W., Turmaine M., Cozens B.A., DiFiglia M., Sharp A.H., Ross C.A., Scherzinger E., Wanker E.E., Mangiarini L., Bates G.P. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. - PubMed
-
- Cummings C.J., Mancini M.A., Antalffy B., DeFranco D.B., Orr H.T., Zoghbi H.Y. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat. Genet. 1998;19:148–154. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
