

VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria
- PMID: 19820692
- PMCID: PMC2799216
- DOI: 10.1038/embor.2009.219
VDAC2 is required for truncated BID-induced mitochondrial apoptosis by recruiting BAK to the mitochondria
Abstract
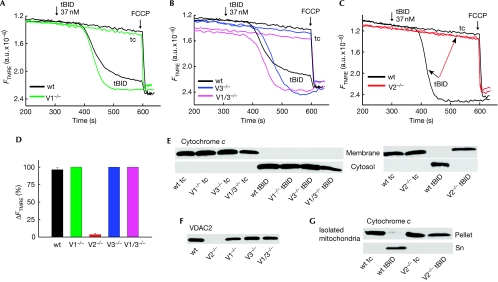
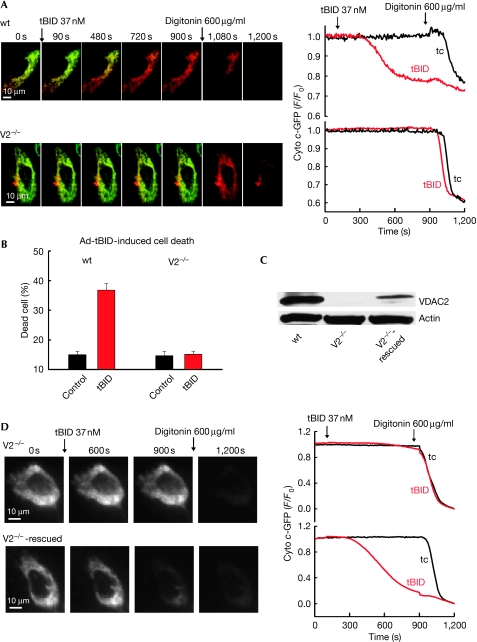
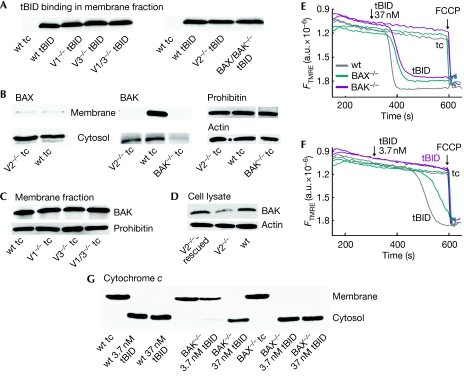
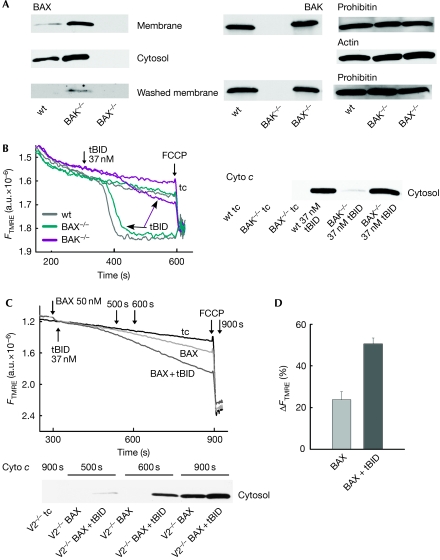
Truncated BID (tBID), a proapoptotic BCL2 family protein, induces BAK/BAX-dependent release of cytochrome c and other mitochondrial intermembrane proteins to the cytosol to induce apoptosis. The voltage-dependent anion channels (VDACs) are the primary gates for solutes across the outer mitochondrial membrane (OMM); however, their role in apoptotic OMM permeabilization remains controversial. Here, we report that VDAC2(-/-) (V2(-/-)) mouse embryonic fibroblasts (MEFs) are virtually insensitive to tBID-induced OMM permeabilization and apoptosis, whereas VDAC1(-/-), VDAC3(-/-) and VDAC1(-/-)/VDAC3(-/-) MEFs respond normally to tBID. V2(-/-) MEFs regain tBID sensitivity after VDAC2 expression. Furthermore, V2(-/-) MEFs are deficient in mitochondrial BAK despite normal tBID-mitochondrial binding and BAX/BAK expression. tBID sensitivity of BAK(-/-) MEFs is also reduced, although not to the same extent as V2(-/-) MEFs, which might result from their strong overexpression of BAX. Indeed, addition of recombinant BAX also sensitized V2(-/-) MEFs to tBID. Thus, VDAC2 acts as a crucial component in mitochondrial apoptosis by allowing the mitochondrial recruitment of BAK, thereby controlling tBID-induced OMM permeabilization and cell death.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Comment in
-
Apoptosis: it's BAK to VDAC.EMBO Rep. 2009 Dec;10(12):1311-3. doi: 10.1038/embor.2009.249. Epub 2009 Nov 13. EMBO Rep. 2009. PMID: 19949413 Free PMC article. Review. No abstract available.
Similar articles
-
Motifs of VDAC2 required for mitochondrial Bak import and tBid-induced apoptosis.Proc Natl Acad Sci U S A. 2015 Oct 13;112(41):E5590-9. doi: 10.1073/pnas.1510574112. Epub 2015 Sep 28. Proc Natl Acad Sci U S A. 2015. PMID: 26417093 Free PMC article.
-
Structure-based modeling of turnover of Bcl-2 family proteins bound to voltage-dependent anion channel 2 (VDAC2): Implications for the mechanisms of proapoptotic activation of Bak and Bax in vivo.Comput Biol Chem. 2020 Apr;85:107203. doi: 10.1016/j.compbiolchem.2020.107203. Epub 2020 Jan 13. Comput Biol Chem. 2020. PMID: 31981967
-
VDAC2 inhibits BAK activation and mitochondrial apoptosis.Science. 2003 Jul 25;301(5632):513-7. doi: 10.1126/science.1083995. Science. 2003. PMID: 12881569
-
Involvement of cardiolipin in tBID-induced activation of BAX during apoptosis.Chem Phys Lipids. 2014 Apr;179:70-4. doi: 10.1016/j.chemphyslip.2013.12.002. Epub 2013 Dec 12. Chem Phys Lipids. 2014. PMID: 24333953 Review.
-
VDAC2-specific cellular functions and the underlying structure.Biochim Biophys Acta. 2016 Oct;1863(10):2503-14. doi: 10.1016/j.bbamcr.2016.04.020. Epub 2016 Apr 23. Biochim Biophys Acta. 2016. PMID: 27116927 Free PMC article. Review.
Cited by
-
Population and single‑cell transcriptome analyses reveal diverse transcriptional changes associated with radioresistance in esophageal squamous cell carcinoma.Int J Oncol. 2019 Dec;55(6):1237-1248. doi: 10.3892/ijo.2019.4897. Epub 2019 Oct 14. Int J Oncol. 2019. PMID: 31638164 Free PMC article.
-
Regulation of the intrinsic apoptosis pathway by reactive oxygen species.Antioxid Redox Signal. 2013 Aug 20;19(6):546-58. doi: 10.1089/ars.2012.4905. Epub 2012 Oct 25. Antioxid Redox Signal. 2013. PMID: 22978471 Free PMC article. Review.
-
Mitochondrial E3 ubiquitin ligase MARCHF5 controls BAK apoptotic activity independently of BH3-only proteins.Cell Death Differ. 2023 Mar;30(3):632-646. doi: 10.1038/s41418-022-01067-z. Epub 2022 Sep 28. Cell Death Differ. 2023. PMID: 36171332 Free PMC article.
-
Mitochondrial Ca(2+) uptake by the voltage-dependent anion channel 2 regulates cardiac rhythmicity.Elife. 2015 Jan 15;4:e04801. doi: 10.7554/eLife.04801. Elife. 2015. PMID: 25588501 Free PMC article.
-
Two roads to death - Bax targets mitochondria by distinct routes before or during apoptotic cell death.Mol Cell Oncol. 2015 Jan 28;2(1):e974460. doi: 10.4161/23723556.2014.974460. eCollection 2015 Jan-Mar. Mol Cell Oncol. 2015. PMID: 27308408 Free PMC article.
References
-
- Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V (2008) Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem 283: 13482–13490 - PubMed
-
- Cheng EH, Sheiko TV, Fisher JK, Craigen WJ, Korsmeyer SJ (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517 - PubMed
-
- Colombini M (2004) VDAC: the channel at the interface between mitochondria and the cytosol. Mol Cell Biochem 256–257: 107–115 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
