

Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability
- PMID: 19783480
- PMCID: PMC2767453
- DOI: 10.1016/j.it.2009.07.012
Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability
Abstract
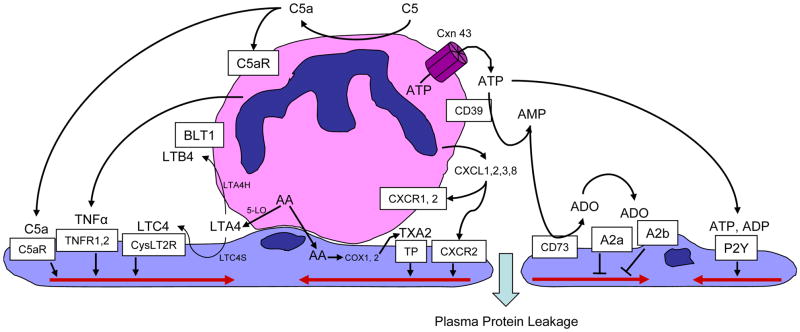
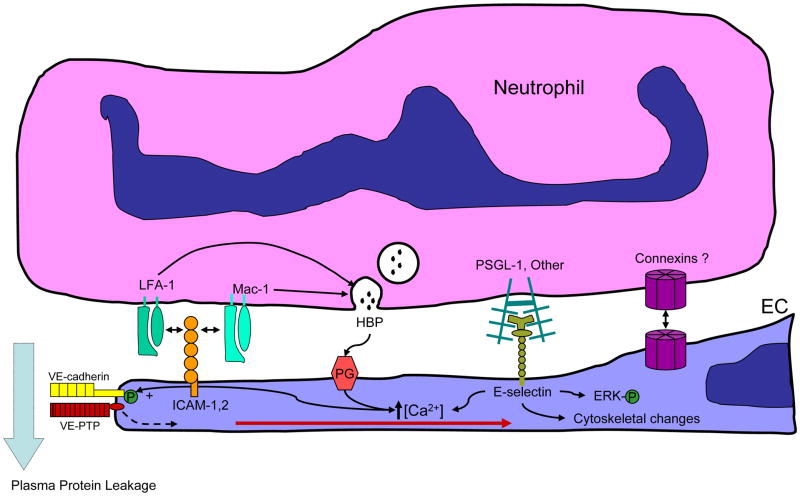
Many diseases have an inflammatory component, where neutrophil interactions with the vascular endothelium lead to barrier dysfunction and increased permeability. Neutrophils increase permeability through secreted products such as the chemokines CXCL1, 2, 3, and 8, through adhesion-dependent processes involving beta(2) integrins interacting with endothelial ICAM-1, and through combinations where beta(2) integrin engagement leads to degranulation and secretion of heparin-binding protein. Some neutrophil products, such as arachidonic acid or the leukotriene LTA4, are further processed by endothelial enzymes via transcellular metabolism before the resulting products thromboxane A2 or LTC4 can activate their cognate receptors. Neutrophils also generate reactive oxygen species that induce vascular leakage. This review focuses on the mechanisms of neutrophil-mediated leakage.
Figures
Similar articles
-
β2 integrin-mediated crawling on endothelial ICAM-1 and ICAM-2 is a prerequisite for transcellular neutrophil diapedesis across the inflamed blood-brain barrier.J Immunol. 2014 Jan 1;192(1):324-37. doi: 10.4049/jimmunol.1300858. Epub 2013 Nov 20. J Immunol. 2014. PMID: 24259506
-
Transcellular biosynthesis of sulfidopeptide leukotrienes during receptor-mediated stimulation of human neutrophil/platelet mixtures.Blood. 1990 Nov 1;76(9):1838-44. Blood. 1990. PMID: 2224131
-
Engagement of ICAM-3 activates polymorphonuclear leukocytes: aggregation without degranulation or beta 2 integrin recruitment.J Immunol. 1998 Dec 1;161(11):6280-7. J Immunol. 1998. PMID: 9834117
-
Integration of inflammatory signals by rolling neutrophils.Immunol Rev. 2002 Aug;186:8-18. doi: 10.1034/j.1600-065x.2002.18602.x. Immunol Rev. 2002. PMID: 12234358 Review.
-
Neutrophil granule proteins tune monocytic cell function.Trends Immunol. 2009 Nov;30(11):538-46. doi: 10.1016/j.it.2009.06.006. Epub 2009 Aug 21. Trends Immunol. 2009. PMID: 19699683 Review.
Cited by
-
Control of stem cell fate and function by engineering physical microenvironments.Integr Biol (Camb). 2012 Sep;4(9):1008-18. doi: 10.1039/c2ib20080e. Integr Biol (Camb). 2012. PMID: 23077731 Free PMC article. Review.
-
Glycolysis, monocarboxylate transport, and purinergic signaling are key events in Eimeria bovis-induced NETosis.Front Immunol. 2022 Aug 11;13:842482. doi: 10.3389/fimmu.2022.842482. eCollection 2022. Front Immunol. 2022. PMID: 36032127 Free PMC article.
-
Myeloperoxidase instigates proinflammatory responses in a cecal ligation and puncture rat model of sepsis.Am J Physiol Heart Circ Physiol. 2020 Sep 1;319(3):H705-H721. doi: 10.1152/ajpheart.00440.2020. Epub 2020 Aug 7. Am J Physiol Heart Circ Physiol. 2020. PMID: 32762560 Free PMC article.
-
Polymorphonuclear neutrophils and instability of the atherosclerotic plaque: a causative role?Inflamm Res. 2013 Jun;62(6):537-50. doi: 10.1007/s00011-013-0617-0. Epub 2013 Apr 3. Inflamm Res. 2013. PMID: 23549741 Review.
-
Adhesion molecules, endothelin-1 and lung function in seven population-based cohorts.Biomarkers. 2013 May;18(3):196-203. doi: 10.3109/1354750X.2012.762805. Epub 2013 Apr 5. Biomarkers. 2013. PMID: 23557128 Free PMC article.
References
-
- Dallegri F, Ottonello L. Tissue injury in neutrophilic inflammation. Inflamm Res. 1997;46 (10):382–391. - PubMed
-
- Lindbom L. Regulation of vascular permeability by neutrophils in acute inflammation. Chem Immunol Allergy. 2003;83:146–166. - PubMed
-
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86 (1):279–367. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
