

Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism
- PMID: 19765191
- PMCID: PMC2766427
- DOI: 10.1111/j.1471-4159.2009.06388.x
Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism
Abstract
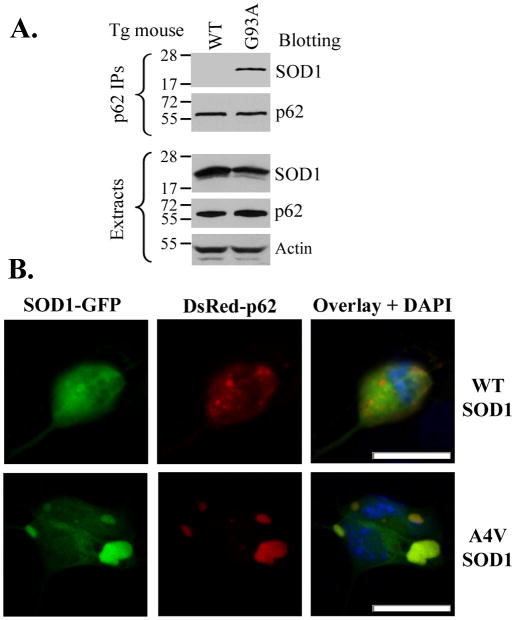
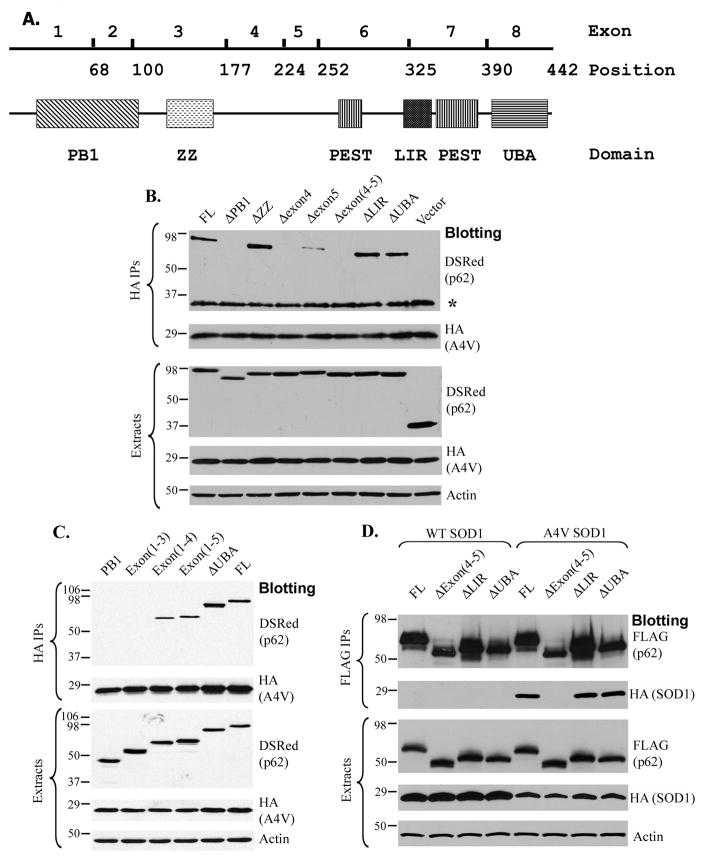
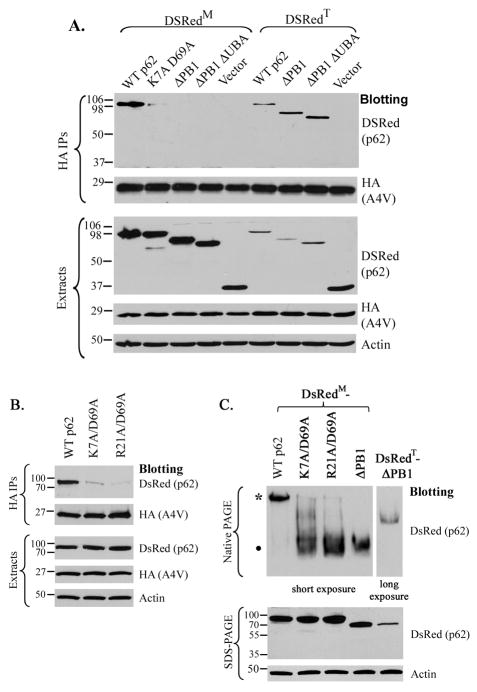
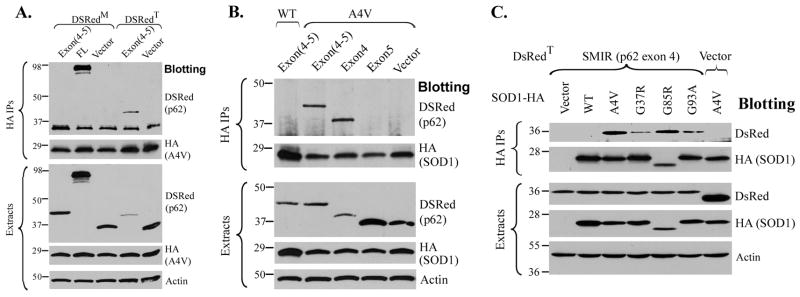
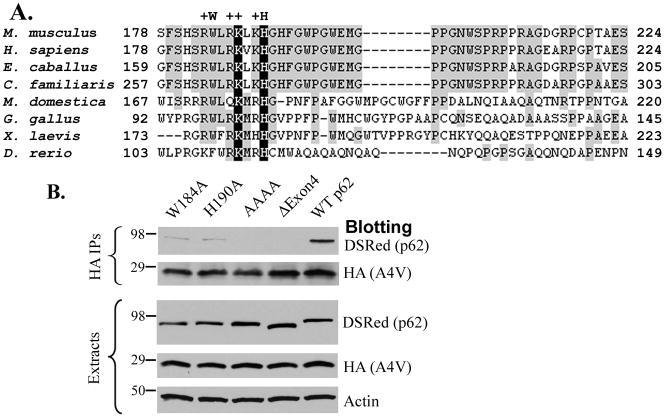
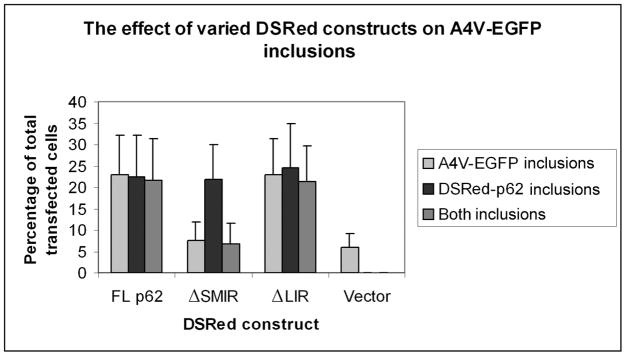
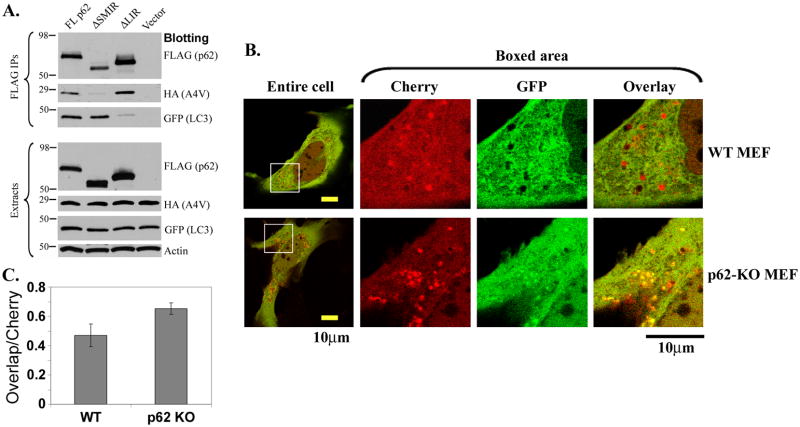
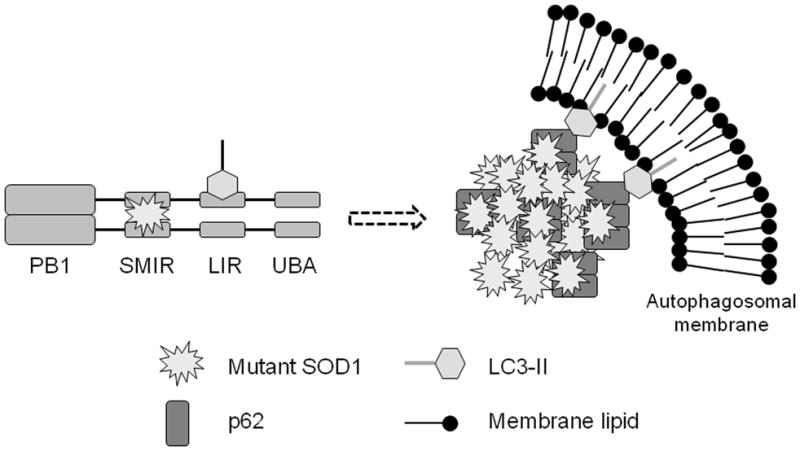
The p62/sequestosome 1 protein has been identified as a component of pathological protein inclusions in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). P62 has also been implicated in autophagy, a process of mass degradation of intracellular proteins and organelles. Autophagy is a critical pathway for degrading misfolded and/or damaged proteins, including the copper-zinc superoxide dismutase (SOD1) mutants linked to familial ALS. We previously reported that p62 interacted with ALS mutants of SOD1 and that the ubiquitin-association domain of p62 was dispensable for the interaction. In this study, we identified two distinct regions of p62 that were essential to its binding to mutant SOD1: the N-terminal Phox and Bem1 (PB1) domain (residues 1-104) and a separate internal region (residues 178-224) termed here as SOD1 mutant interaction region (SMIR). The PB1 domain is required for appropriate oligomeric status of p62 and the SMIR is the actual region interacting with mutant SOD1. Within the SMIR, the conserved W184, H190 and positively charged R183, R186, K187, and K189 residues are critical to the p62-mutant SOD1 interaction as substitution of these residues with alanine resulted in significantly abolished binding. In addition, SMIR and the p62 sequence responsible for the interaction with LC3, a protein essential for autophagy activation, are independent of each other. In cells lacking p62, the existence of mutant SOD1 in acidic autolysosomes decreased, suggesting that p62 can function as an adaptor between mutant SOD1 and the autophagy machinery. This study provides a novel molecular mechanism by which mutant SOD1 can be recognized by p62 in an ubiquitin-independent fashion and targeted for the autophagy-lysosome degradation pathway.
Figures
Similar articles
-
p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis.J Biol Chem. 2007 Apr 13;282(15):11068-77. doi: 10.1074/jbc.M608787200. Epub 2007 Feb 12. J Biol Chem. 2007. PMID: 17296612
-
Absence of lipofuscin in motor neurons of SOD1-linked ALS mice.Proc Natl Acad Sci U S A. 2014 Jul 29;111(30):11055-60. doi: 10.1073/pnas.1409314111. Epub 2014 Jul 14. Proc Natl Acad Sci U S A. 2014. PMID: 25024188 Free PMC article.
-
The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS).Hum Mol Genet. 2010 Sep 1;19(17):3440-56. doi: 10.1093/hmg/ddq257. Epub 2010 Jun 22. Hum Mol Genet. 2010. PMID: 20570967
-
Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response.Prog Neurobiol. 2012 May;97(2):101-26. doi: 10.1016/j.pneurobio.2011.10.001. Epub 2011 Oct 20. Prog Neurobiol. 2012. PMID: 22033150 Review.
-
p62 Stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress.Trends Cardiovasc Med. 2011 Nov;21(8):224-8. doi: 10.1016/j.tcm.2012.05.015. Trends Cardiovasc Med. 2011. PMID: 22902070 Free PMC article. Review.
Cited by
-
ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway.Autophagy. 2020 May;16(5):917-931. doi: 10.1080/15548627.2019.1644076. Epub 2019 Jul 30. Autophagy. 2020. PMID: 31362587 Free PMC article.
-
Manipulation or capitulation: virus interactions with autophagy.Microbes Infect. 2012 Feb;14(2):126-39. doi: 10.1016/j.micinf.2011.09.007. Epub 2011 Oct 24. Microbes Infect. 2012. PMID: 22051604 Free PMC article. Review.
-
TDP-43 and Inflammation: Implications for Amyotrophic Lateral Sclerosis and Frontotemporal Dementia.Int J Mol Sci. 2021 Jul 21;22(15):7781. doi: 10.3390/ijms22157781. Int J Mol Sci. 2021. PMID: 34360544 Free PMC article. Review.
-
Amyotrophic lateral sclerosis patient iPSC-derived astrocytes impair autophagy via non-cell autonomous mechanisms.Mol Brain. 2017 Jun 13;10(1):22. doi: 10.1186/s13041-017-0300-4. Mol Brain. 2017. PMID: 28610619 Free PMC article.
-
Autophagy dysregulation in amyotrophic lateral sclerosis.Brain Pathol. 2012 Jan;22(1):110-6. doi: 10.1111/j.1750-3639.2011.00546.x. Brain Pathol. 2012. PMID: 22150926 Free PMC article. Review.
References
-
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. - PubMed
-
- Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget's disease of bone. J Biol Chem. 2003;278:37409–37412. - PubMed
-
- Denk H, Stumptner C, Fuchsbichler A, Muller T, Farr G, Muller W, Terracciano L, Zatloukal K. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J Pathol. 2006;208:653–661. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P20 RR020171/RR/NCRR NIH HHS/United States
- P42 ES007380-129005/ES/NIEHS NIH HHS/United States
- P42 ES007380/ES/NIEHS NIH HHS/United States
- R21 AG032567-01/AG/NIA NIH HHS/United States
- P20 RR020171-057026/RR/NCRR NIH HHS/United States
- R21 AG032567/AG/NIA NIH HHS/United States
- R01NS049126/NS/NINDS NIH HHS/United States
- R01 MH098891/MH/NIMH NIH HHS/United States
- P42ES007380/ES/NIEHS NIH HHS/United States
- R01 NS049126/NS/NINDS NIH HHS/United States
- P42 ES007380-139005/ES/NIEHS NIH HHS/United States
- R21AG032567/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
