

The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia
- PMID: 19732723
- PMCID: PMC2931825
- DOI: 10.1016/j.ccr.2009.07.030
The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia
Abstract
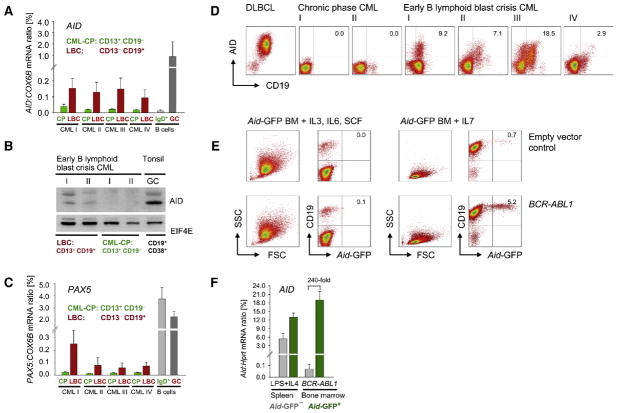
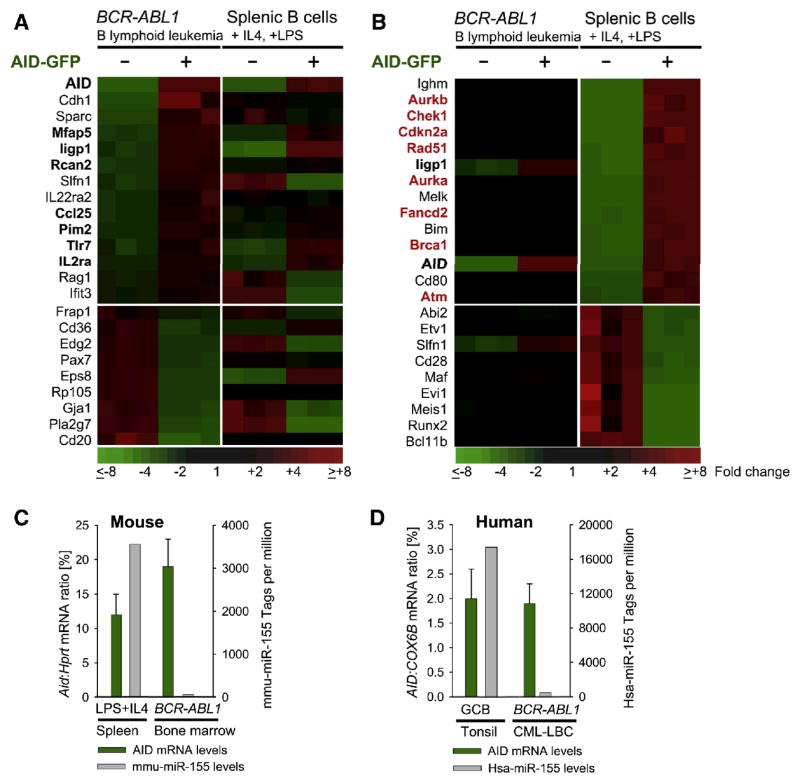
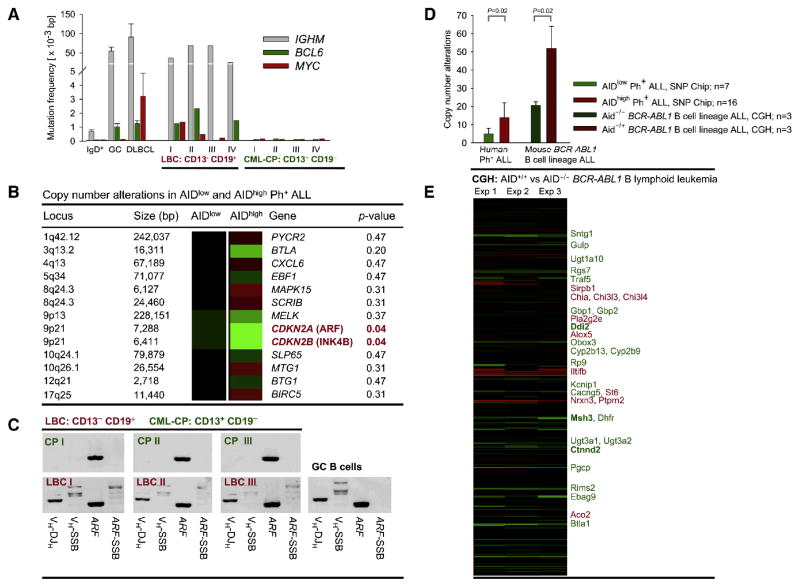
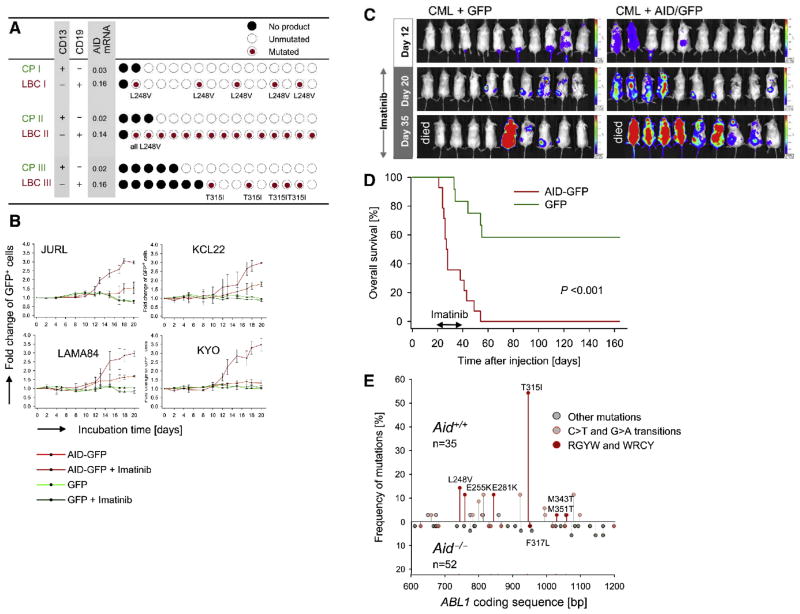
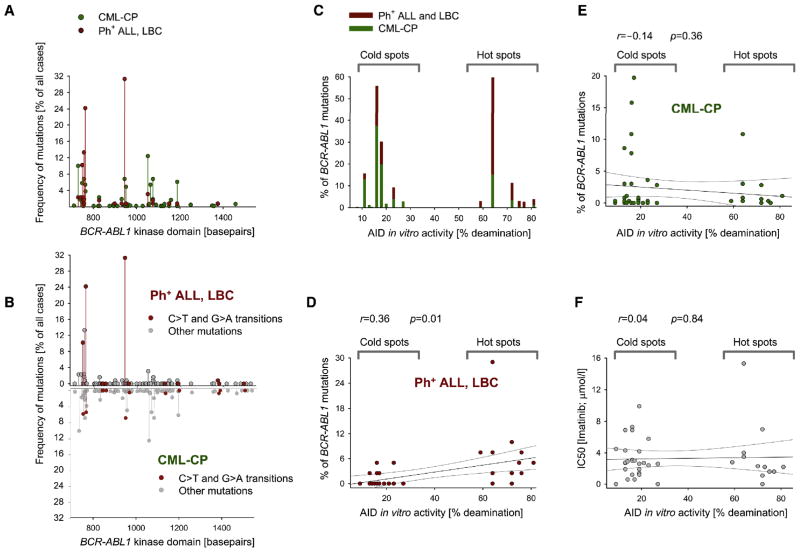
Chronic myeloid leukemia (CML) is induced by BCR-ABL1 and can be effectively treated for many years with Imatinib until leukemia cells acquire drug resistance through BCR-ABL1 mutations and progress into fatal B lymphoid blast crisis (LBC). Despite its clinical significance, the mechanism of progression into LBC is unknown. Here, we show that LBC but not CML cells express the B cell-specific mutator enzyme AID. We demonstrate that AID expression in CML cells promotes overall genetic instability by hypermutation of tumor suppressor and DNA repair genes. Importantly, our data uncover a causative role of AID activity in the acquisition of BCR-ABL1 mutations leading to Imatinib resistance, thus providing a rationale for the rapid development of drug resistance and blast crisis progression.
Conflict of interest statement
The authors have no conflicting financial interests.
Figures

Comment in
-
Imatinib resistance and progression of CML to blast crisis: somatic hypermutation AIDing the way.Cancer Cell. 2009 Sep 8;16(3):174-6. doi: 10.1016/j.ccr.2009.08.012. Cancer Cell. 2009. PMID: 19732715
Similar articles
-
Imatinib resistance and progression of CML to blast crisis: somatic hypermutation AIDing the way.Cancer Cell. 2009 Sep 8;16(3):174-6. doi: 10.1016/j.ccr.2009.08.012. Cancer Cell. 2009. PMID: 19732715
-
AID expression is correlated with Bcr-Abl expression in CML-LBC and can be down-regulated by As2O3 and/or imatinib.Leuk Res. 2011 Oct;35(10):1355-9. doi: 10.1016/j.leukres.2011.04.020. Epub 2011 May 13. Leuk Res. 2011. PMID: 21570118
-
[Research advance on molecular genetics of CML blast crisis].Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2008 Feb;16(1):217-21. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2008. PMID: 18315935 Review. Chinese.
-
Triptolide induces cell death independent of cellular responses to imatinib in blast crisis chronic myelogenous leukemia cells including quiescent CD34+ primitive progenitor cells.Mol Cancer Ther. 2009 Sep;8(9):2509-16. doi: 10.1158/1535-7163.MCT-09-0386. Epub 2009 Sep 1. Mol Cancer Ther. 2009. PMID: 19723894 Free PMC article.
-
Resistance in the land of molecular cancer therapeutics.Cancer Cell. 2002 Aug;2(2):99-102. doi: 10.1016/s1535-6108(02)00101-0. Cancer Cell. 2002. PMID: 12204529 Review.
Cited by
-
RAG enhances BCR-ABL1-positive leukemic cell growth through its endonuclease activity in vitro and in vivo.Cancer Sci. 2021 Jul;112(7):2679-2691. doi: 10.1111/cas.14939. Epub 2021 May 18. Cancer Sci. 2021. PMID: 33949040 Free PMC article.
-
ABL fusion oncogene transformation and inhibitor sensitivity are mediated by the cellular regulator RIN1.Leukemia. 2011 Feb;25(2):290-300. doi: 10.1038/leu.2010.268. Epub 2010 Nov 19. Leukemia. 2011. PMID: 21102429 Free PMC article.
-
Concomitant AID Expression and BCL7A Loss Associates With Accelerated Phase Progression and Imatinib Resistance in Chronic Myeloid Leukemia.Ann Lab Med. 2017 Mar;37(2):177-179. doi: 10.3343/alm.2017.37.2.177. Ann Lab Med. 2017. PMID: 28029010 Free PMC article. No abstract available.
-
CDKN2A-independent role of BMI1 in promoting growth and survival of Ph+ acute lymphoblastic leukemia.Leukemia. 2016 Aug;30(8):1682-90. doi: 10.1038/leu.2016.70. Epub 2016 Apr 5. Leukemia. 2016. PMID: 27125204 Free PMC article.
-
Activation-induced cytidine deaminase accelerates clonal evolution in BCR-ABL1-driven B-cell lineage acute lymphoblastic leukemia.Cancer Res. 2010 Oct 1;70(19):7411-20. doi: 10.1158/0008-5472.CAN-10-1438. Epub 2010 Sep 28. Cancer Res. 2010. PMID: 20876806 Free PMC article.
References
-
- Calabretta B, Perrotti D. The biology of CML blast crisis. Blood. 2004;103:4010–22. - PubMed
-
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. - PubMed
-
- Chesi M, Robbiani DF, Sebag M, Chang WJ, Affer M, Tiedemann R, Valdez R, Palmer SE, Haas SS, Stewart AK, Fonseca R, et al. AID-dependent activation of a MYC transgene induces multiple myeloma in a conditional mouse model of post-germinal center malignancies. Cancer Cell. 2008;13:167–80. - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
