

Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis
- PMID: 19653905
- PMCID: PMC2731755
- DOI: 10.1186/1471-2105-10-239
Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis
Abstract
Background: Transcriptome sequences provide a complement to structural genomic information and provide snapshots of an organism's transcriptional profile. Such sequences also represent an alternative method for characterizing neglected species that are not expected to undergo whole-genome sequencing. One difficulty for transcriptome sequencing of these organisms is the low quality of reads and incomplete coverage of transcripts, both of which compromise further bioinformatics analyses. Another complicating factor is the lack of known protein homologs, which frustrates searches against established protein databases. This lack of homologs may be caused by divergence from well-characterized and over-represented model organisms. Another explanation is that non-coding RNAs (ncRNAs) may be caught during sequencing. NcRNAs are RNA sequences that, unlike messenger RNAs, do not code for protein products and instead perform unique functions by folding into higher order structural conformations. There is ncRNA screening software available that is specific for transcriptome sequences, but their analyses are optimized for those transcriptomes that are well represented in protein databases, and also assume that input ESTs are full-length and high quality.
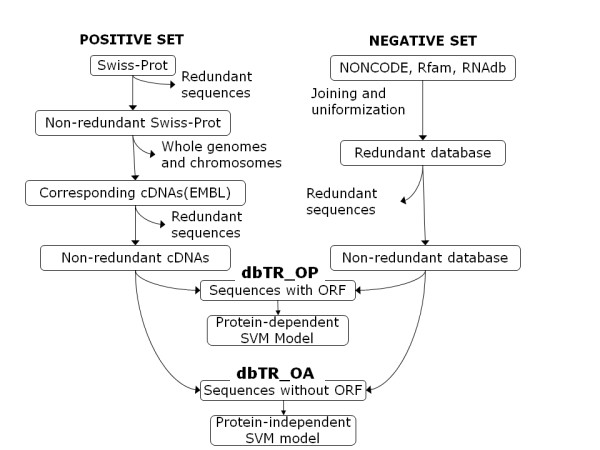
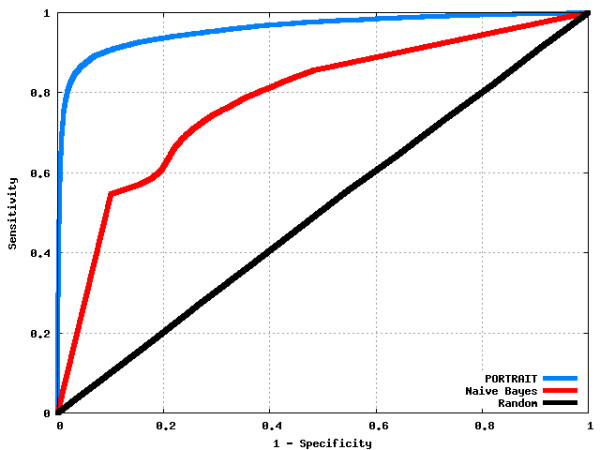
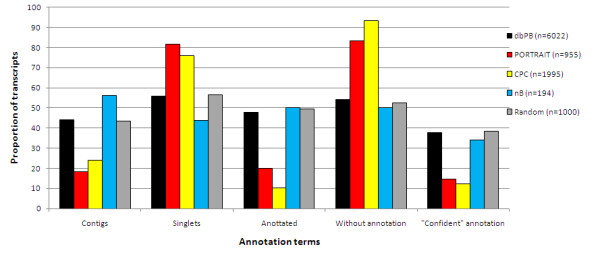
Results: We propose an algorithm called PORTRAIT, which is suitable for ncRNA analysis of transcriptomes from poorly characterized species. Sequences are translated by software that is resistant to sequencing errors, and the predicted putative proteins, along with their source transcripts, are evaluated for coding potential by a support vector machine (SVM). Either of two SVM models may be employed: if a putative protein is found, a protein-dependent SVM model is used; if it is not found, a protein-independent SVM model is used instead. Only ab initio features are extracted, so that no homology information is needed. We illustrate the use of PORTRAIT by predicting ncRNAs from the transcriptome of the pathogenic fungus Paracoccidoides brasiliensis and five other related fungi.
Conclusion: PORTRAIT can be integrated into pipelines, and provides a low computational cost solution for ncRNA detection in transcriptome sequencing projects.
Figures
Similar articles
-
Transcriptomic profiling identifies novel transcripts, isomorphs, and noncoding RNAs in Paracoccidioides brasiliensis.Med Mycol. 2021 Feb 4;59(2):197-200. doi: 10.1093/mmy/myaa062. Med Mycol. 2021. PMID: 32692810
-
The transcriptome analysis of early morphogenesis in Paracoccidioides brasiliensis mycelium reveals novel and induced genes potentially associated to the dimorphic process.BMC Microbiol. 2007 Apr 10;7:29. doi: 10.1186/1471-2180-7-29. BMC Microbiol. 2007. PMID: 17425801 Free PMC article.
-
Knowledge-based reasoning to annotate noncoding RNA using multi-agent system.J Bioinform Comput Biol. 2015 Dec;13(6):1550021. doi: 10.1142/S0219720015500213. Epub 2015 Jun 24. J Bioinform Comput Biol. 2015. PMID: 26223200
-
Identification and annotation of noncoding RNAs in Saccharomycotina.C R Biol. 2011 Aug-Sep;334(8-9):671-8. doi: 10.1016/j.crvi.2011.05.016. Epub 2011 Jul 6. C R Biol. 2011. PMID: 21819949 Review.
-
Computational methods for annotation of plant regulatory non-coding RNAs using RNA-seq.Brief Bioinform. 2021 Jul 20;22(4):bbaa322. doi: 10.1093/bib/bbaa322. Brief Bioinform. 2021. PMID: 33333550 Review.
Cited by
-
Long non-coding RNA-encoded micropeptides: functions, mechanisms and implications.Cell Death Discov. 2024 Oct 23;10(1):450. doi: 10.1038/s41420-024-02175-0. Cell Death Discov. 2024. PMID: 39443468 Free PMC article. Review.
-
Common Features in lncRNA Annotation and Classification: A Survey.Noncoding RNA. 2021 Dec 13;7(4):77. doi: 10.3390/ncrna7040077. Noncoding RNA. 2021. PMID: 34940758 Free PMC article. Review.
-
Identification and Expression Analysis of Long Noncoding RNAs in Fat-Tail of Sheep Breeds.G3 (Bethesda). 2019 Apr 9;9(4):1263-1276. doi: 10.1534/g3.118.201014. G3 (Bethesda). 2019. PMID: 30787031 Free PMC article.
-
Exosomes Could Offer New Options to Combat the Long-Term Complications Inflicted by Gestational Diabetes Mellitus.Cells. 2020 Mar 10;9(3):675. doi: 10.3390/cells9030675. Cells. 2020. PMID: 32164322 Free PMC article. Review.
-
De novo transcriptome assembly from inflorescence of Orchis italica: analysis of coding and non-coding transcripts.PLoS One. 2014 Jul 15;9(7):e102155. doi: 10.1371/journal.pone.0102155. eCollection 2014. PLoS One. 2014. PMID: 25025767 Free PMC article.
References
-
- Ravasi T, Suzuki H, Pang KC, Katayama S, Furuno M, Okunishi R, Fukuda S, Ru K, Frith MC, Gongora MM, Grimmond SM, Hume DA, Hayashizaki Y, Mattick JS. Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 2006;16:11–19. doi: 10.1101/gr.4200206. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
