

Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice
- PMID: 19652361
- PMCID: PMC2735933
- DOI: 10.1172/JCI39162
Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice
Abstract
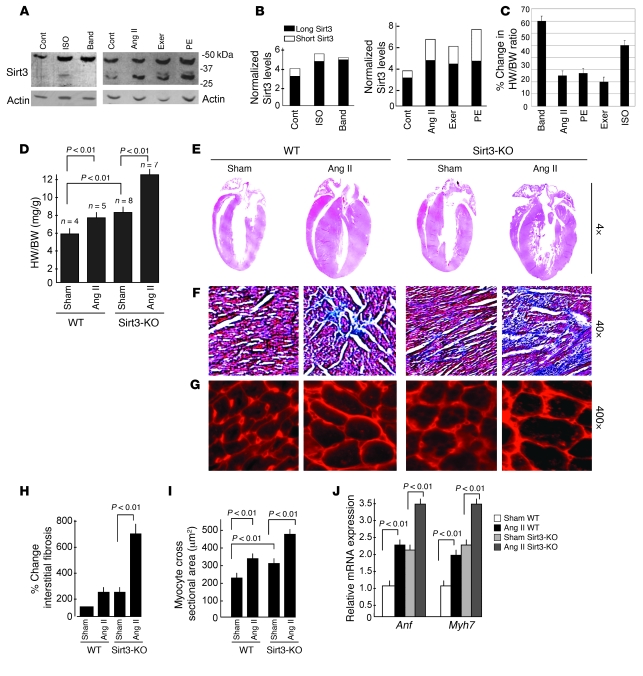
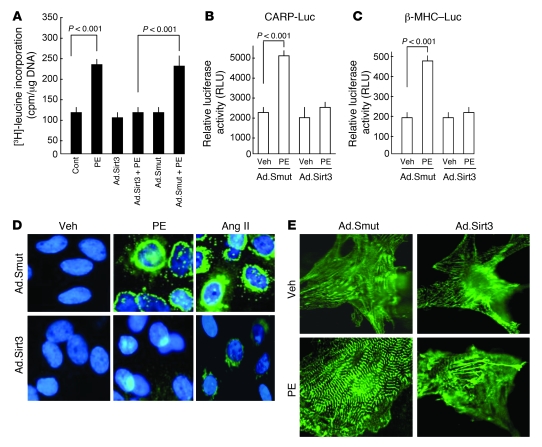
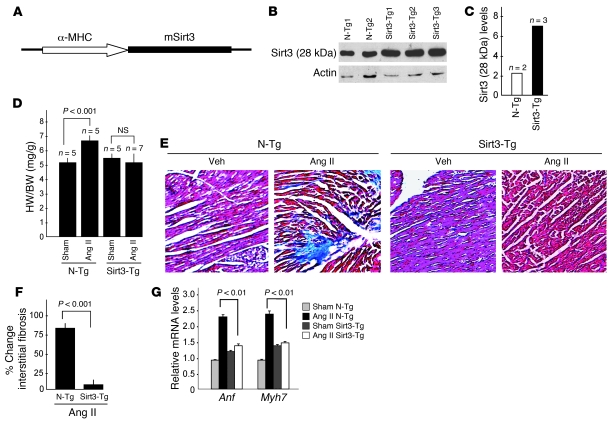
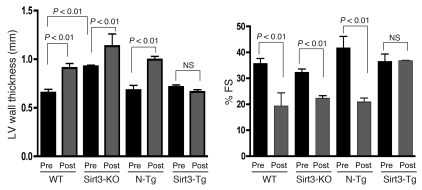
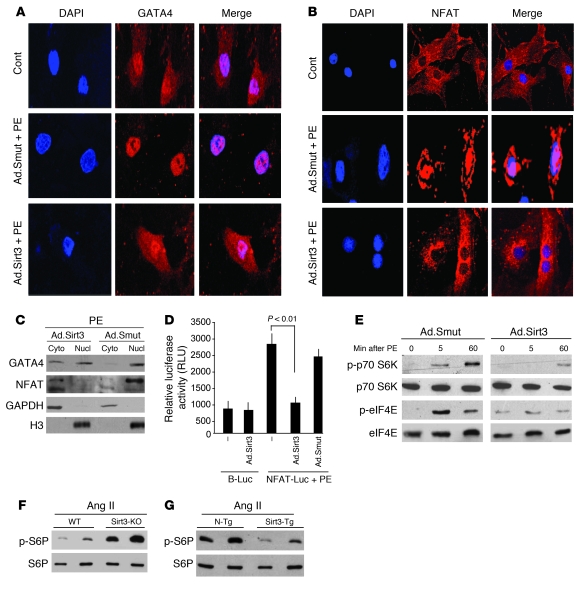
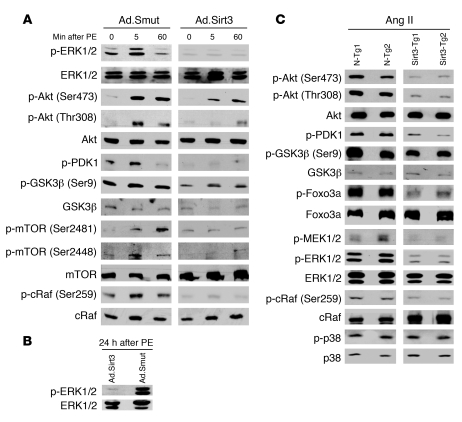
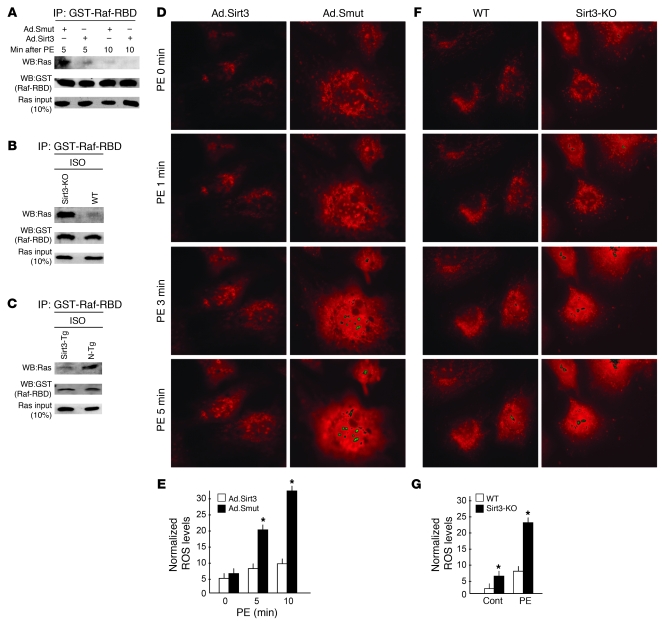
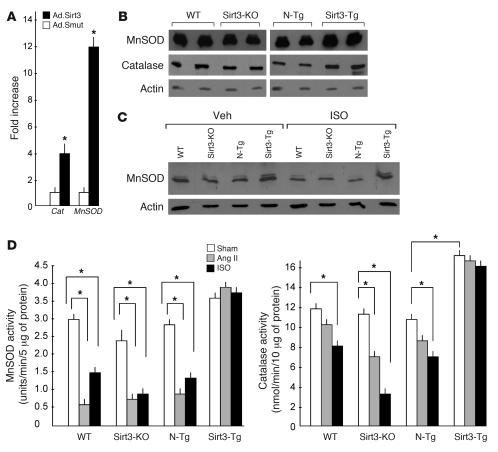
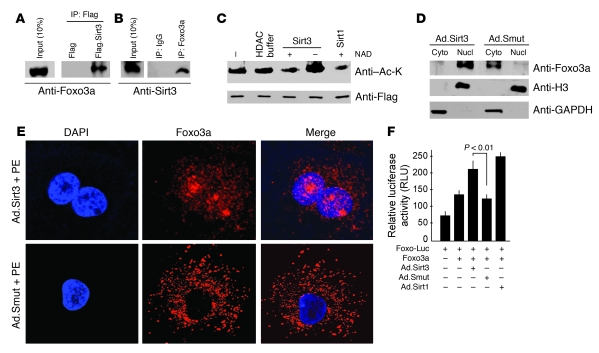
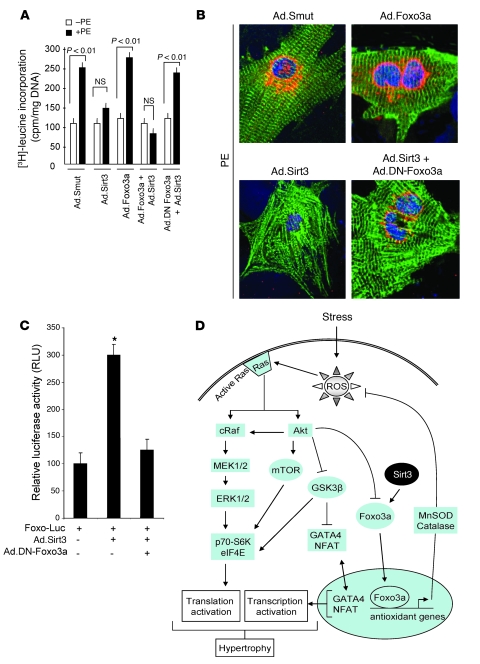
Sirtuin 3 (SIRT3) is a member of the sirtuin family of proteins that promote longevity in many organisms. Increased expression of SIRT3 has been linked to an extended life span in humans. Here, we have shown that Sirt3 protects the mouse heart by blocking the cardiac hypertrophic response. Although Sirt3-deficient mice appeared to have normal activity, they showed signs of cardiac hypertrophy and interstitial fibrosis at 8 weeks of age. Application of hypertrophic stimuli to these mice produced a severe cardiac hypertrophic response, whereas Sirt3-expressing Tg mice were protected from similar stimuli. In primary cultures of cardiomyocytes, Sirt3 blocked cardiac hypertrophy by activating the forkhead box O3a-dependent (Foxo3a-dependent), antioxidant-encoding genes manganese superoxide dismutase (MnSOD) and catalase (Cat), thereby decreasing cellular levels of ROS. Reduced ROS levels suppressed Ras activation and downstream signaling through the MAPK/ERK and PI3K/Akt pathways. This resulted in repressed activity of transcription factors, specifically GATA4 and NFAT, and translation factors, specifically eukaryotic initiation factor 4E (elf4E) and S6 ribosomal protein (S6P), which are involved in the development of cardiac hypertrophy. These results demonstrate that SIRT3 is an endogenous negative regulator of cardiac hypertrophy, which protects hearts by suppressing cellular levels of ROS.
Figures
Similar articles
-
Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia.Neuroscience. 2015 Dec 17;311:398-414. doi: 10.1016/j.neuroscience.2015.10.048. Epub 2015 Oct 30. Neuroscience. 2015. PMID: 26523980
-
SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression.Int J Biol Sci. 2008 Sep 5;4(5):291-9. doi: 10.7150/ijbs.4.291. Int J Biol Sci. 2008. PMID: 18781224 Free PMC article.
-
SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms.Basic Res Cardiol. 2016 Mar;111(2):13. doi: 10.1007/s00395-016-0531-z. Epub 2016 Jan 19. Basic Res Cardiol. 2016. PMID: 26786260
-
The role of sirtuins in cardiac disease.Am J Physiol Heart Circ Physiol. 2015 Nov;309(9):H1375-89. doi: 10.1152/ajpheart.00053.2015. Epub 2015 Jul 31. Am J Physiol Heart Circ Physiol. 2015. PMID: 26232232 Free PMC article. Review.
-
Mitochondrial SIRT3 and heart disease.Cardiovasc Res. 2010 Nov 1;88(2):250-6. doi: 10.1093/cvr/cvq250. Epub 2010 Aug 4. Cardiovasc Res. 2010. PMID: 20685942 Free PMC article. Review.
Cited by
-
Histone Deacetylase 3 (HDAC3)-dependent Reversible Lysine Acetylation of Cardiac Myosin Heavy Chain Isoforms Modulates Their Enzymatic and Motor Activity.J Biol Chem. 2015 Jun 19;290(25):15559-15569. doi: 10.1074/jbc.M115.653048. Epub 2015 Apr 24. J Biol Chem. 2015. PMID: 25911107 Free PMC article.
-
Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association.Circ Res. 2016 Jun 10;118(12):1960-91. doi: 10.1161/RES.0000000000000104. Epub 2016 Apr 28. Circ Res. 2016. PMID: 27126807 Free PMC article. Review.
-
SIRT3 is attenuated in systemic sclerosis skin and lungs, and its pharmacologic activation mitigates organ fibrosis.Oncotarget. 2016 Oct 25;7(43):69321-69336. doi: 10.18632/oncotarget.12504. Oncotarget. 2016. PMID: 27732568 Free PMC article.
-
Exploring Sirtuins: New Frontiers in Managing Heart Failure with Preserved Ejection Fraction.Int J Mol Sci. 2024 Jul 15;25(14):7740. doi: 10.3390/ijms25147740. Int J Mol Sci. 2024. PMID: 39062982 Free PMC article. Review.
-
The association between left ventricular mass index and serum sirtuin 3 level in patients with hypertension.Cardiovasc Endocrinol Metab. 2020 Aug 27;10(2):99-105. doi: 10.1097/XCE.0000000000000231. eCollection 2021 Jun. Cardiovasc Endocrinol Metab. 2020. PMID: 34113795 Free PMC article.
References
-
- Frey N., Katus H.A., Olson E.N., Hill J.A. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB. - DOI - PubMed
-
- Heineke J., Molkentin J.D. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat. Rev. Mol. Cell Biol. 2006;7:589–600. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
