

Activation of SIRT1 by resveratrol represses transcription of the gene for the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) by deacetylating hepatic nuclear factor 4alpha
- PMID: 19651778
- PMCID: PMC2785634
- DOI: 10.1074/jbc.M109.047340
Activation of SIRT1 by resveratrol represses transcription of the gene for the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) by deacetylating hepatic nuclear factor 4alpha
Abstract
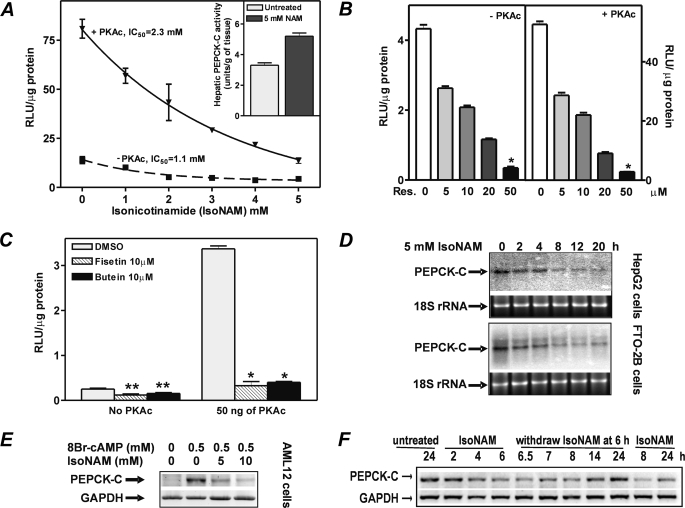
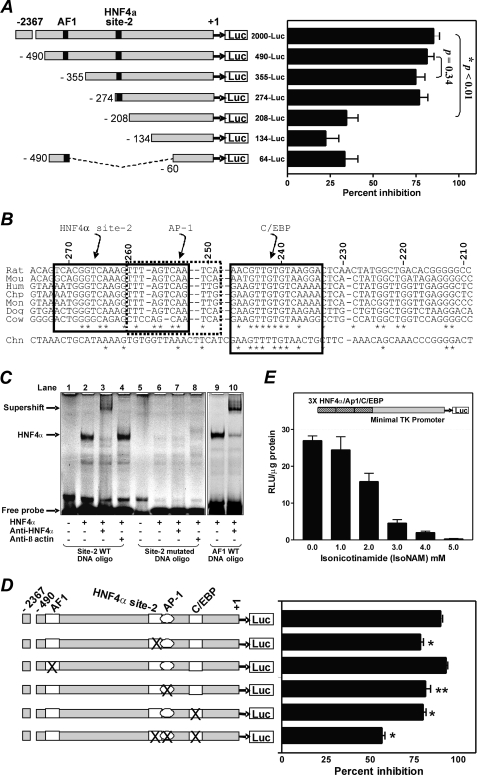
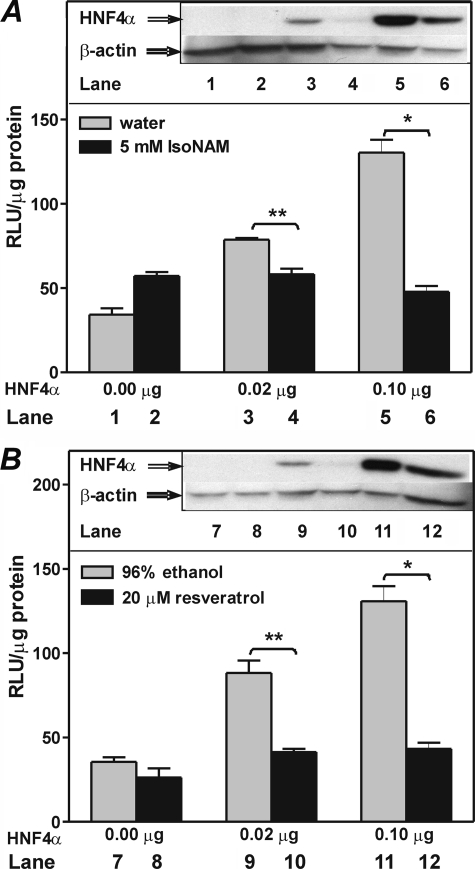
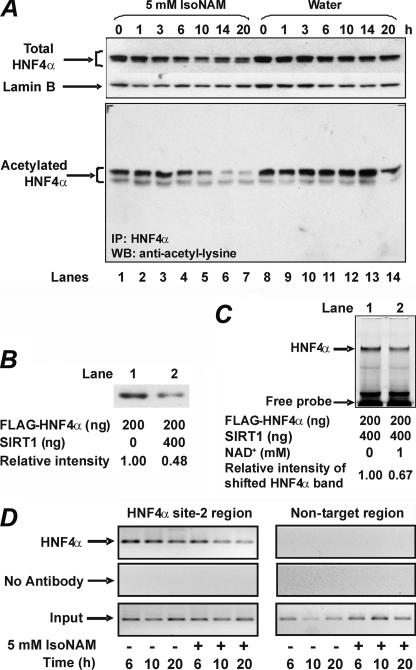
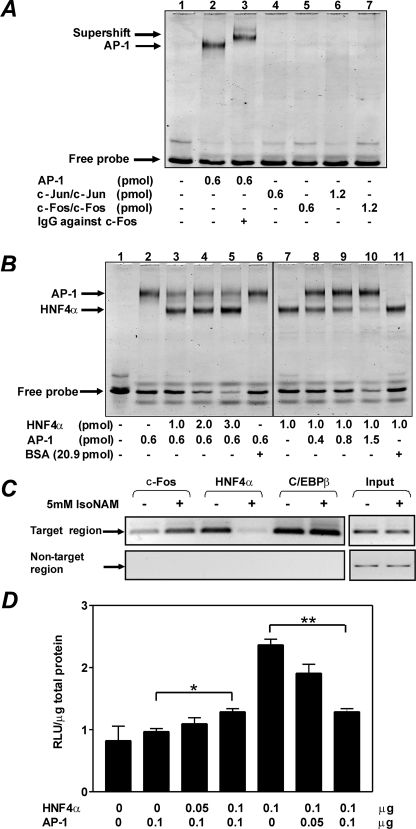
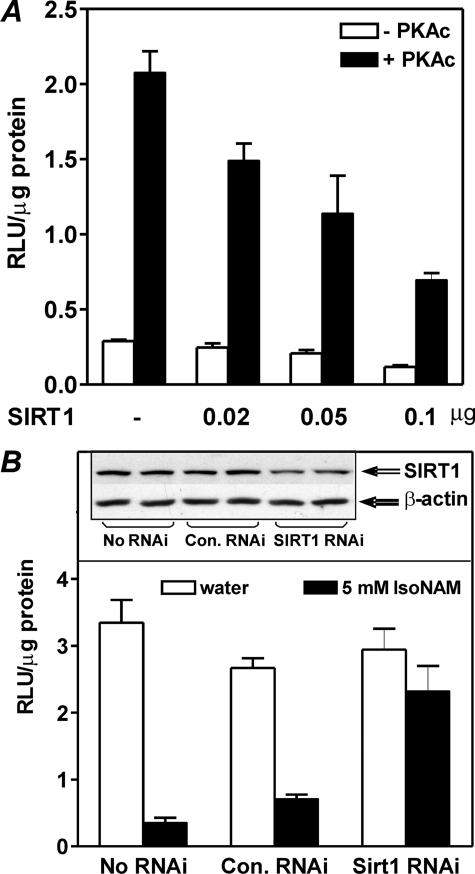
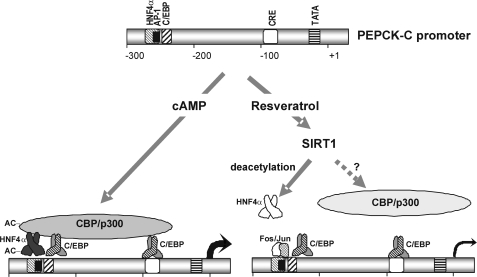
The SIRT1 activators isonicotinamide (IsoNAM), resveratrol, fisetin, and butein repressed transcription of the gene for the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) (PEPCK-C). An evolutionarily conserved binding site for hepatic nuclear factor (HNF) 4alpha (-272/-252) was identified, which was required for transcriptional repression of the PEPCK-C gene promoter caused by these compounds. This site contains an overlapping AP-1 binding site and is adjacent to the C/EBP binding element (-248/-234); the latter is necessary for hepatic transcription of PEPCK-C. AP-1 competed with HNF4alpha for binding to this site and also decreased HNF4alpha stimulation of transcription from the PEPCK-C gene promoter. Chromatin immunoprecipitation experiments demonstrated that HNF4alpha and AP-1, but not C/EBPbeta, reciprocally bound to this site prior to and after treating HepG2 cells with IsoNAM. IsoNAM treatment resulted in deacetylation of HNF4alpha, which decreased its binding affinity to the PEPCK-C gene promoter. In HNF4alpha-null Chinese hamster ovary cells, IsoNAM and resveratrol failed to repress transcription from the PEPCK-C gene promoter; overexpression of HNF4alpha in Chinese hamster ovary cells re-established transcriptional inhibition. Exogenous SIRT1 expression repressed transcription, whereas knockdown of SIRT1 by RNA interference reversed this effect. IsoNAM decreased the level of mRNA for PEPCK-C but had no effect on mRNA for glucose-6-phosphatase in AML12 mouse hepatocytes. We conclude that SIRT1 activation inhibited transcription of the gene for PEPCK-C in part by deacetylation of HNF4alpha. However, SIRT1 deacetylation of other key regulatory proteins that control PEPCK-C gene transcription also likely contributed to the inhibitory effect.
Figures
Similar articles
-
Glucocorticoids repress transcription of phosphoenolpyruvate carboxykinase (GTP) gene in adipocytes by inhibiting its C/EBP-mediated activation.J Biol Chem. 2003 Apr 11;278(15):12929-36. doi: 10.1074/jbc.M300263200. Epub 2003 Jan 30. J Biol Chem. 2003. PMID: 12560325
-
The transcriptional regulation of phosphoenolpyruvate carboxykinase gene in the kidney requires the HNF-1 binding site of the gene.Gene. 2003 Oct 30;318:177-84. doi: 10.1016/s0378-1119(03)00775-3. Gene. 2003. PMID: 14585510
-
Relative roles of CCAAT/enhancer-binding protein beta and cAMP regulatory element-binding protein in controlling transcription of the gene for phosphoenolpyruvate carboxykinase (GTP).J Biol Chem. 1993 Jan 5;268(1):613-9. J Biol Chem. 1993. PMID: 8093246
-
Cell-specific expression of cytosolic phosphoenolpyruvate carboxykinase in transgenic mice.FASEB J. 1992 Dec;6(15):3330-7. doi: 10.1096/fasebj.6.15.1281456. FASEB J. 1992. PMID: 1281456 Review.
-
Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression.Annu Rev Biochem. 1997;66:581-611. doi: 10.1146/annurev.biochem.66.1.581. Annu Rev Biochem. 1997. PMID: 9242918 Review.
Cited by
-
Resveratrol ameliorates imiquimod-induced psoriasis-like skin inflammation in mice.PLoS One. 2015 May 12;10(5):e0126599. doi: 10.1371/journal.pone.0126599. eCollection 2015. PLoS One. 2015. PMID: 25965695 Free PMC article.
-
Sirtuin 1 (SIRT1) protein degradation in response to persistent c-Jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity.J Biol Chem. 2011 Jun 24;286(25):22227-34. doi: 10.1074/jbc.M111.228874. Epub 2011 May 3. J Biol Chem. 2011. PMID: 21540183 Free PMC article.
-
Enhancing glucose metabolism via gluconeogenesis is therapeutic in a zebrafish model of Dravet syndrome.Brain Commun. 2021 Jan 25;3(1):fcab004. doi: 10.1093/braincomms/fcab004. eCollection 2021. Brain Commun. 2021. PMID: 33842883 Free PMC article.
-
Feedback regulation of hepatic gluconeogenesis through modulation of SHP/Nr0b2 gene expression by Sirt1 and FoxO1.Am J Physiol Endocrinol Metab. 2011 Feb;300(2):E312-20. doi: 10.1152/ajpendo.00524.2010. Epub 2010 Nov 16. Am J Physiol Endocrinol Metab. 2011. PMID: 21081708 Free PMC article.
-
Supplementing media with NAD+ precursors enhances the in vitro maturation of porcine oocytes.J Reprod Dev. 2021 Oct 29;67(5):319-326. doi: 10.1262/jrd.2021-080. Epub 2021 Aug 19. J Reprod Dev. 2021. PMID: 34408103 Free PMC article.
References
-
- Hanson R. W., Patel Y. M. (eds) (1994) P-enolpyruvate Carboxykinase: The Gene and the Enzyme, John Wiley and Sons, New York
-
- Franckhauser S., Muñoz S., Pujol A., Casellas A., Riu E., Otaegui P., Su B., Bosch F. (2002) Diabetes 51, 624–630 - PubMed
-
- Franckhauser S., Muñoz S., Elias I., Ferre T., Bosch F. (2006) Diabetes 55, 273–280 - PubMed
-
- Reshef L., Olswang Y., Cassuto H., Blum B., Croniger C. M., Kalhan S. C., Tilghman S. M., Hanson R. W. (2003) J. Biol. Chem. 278, 30413–30416 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
