

Ligands, their receptors and ... plasma membranes
- PMID: 19647036
- PMCID: PMC7116919
- DOI: 10.1016/j.mce.2009.07.022
Ligands, their receptors and ... plasma membranes
Abstract
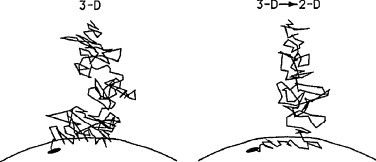
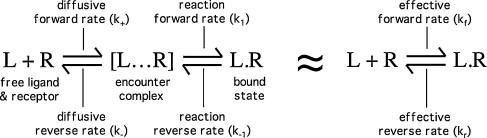
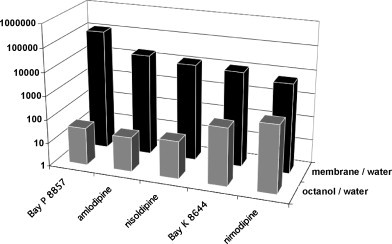
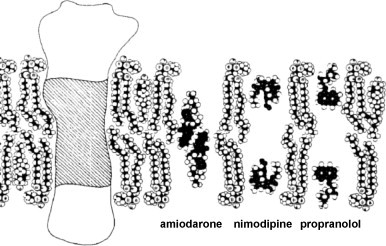
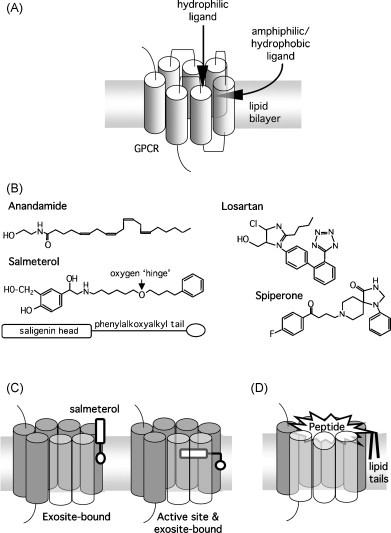
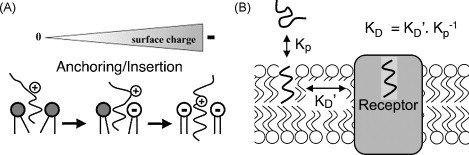
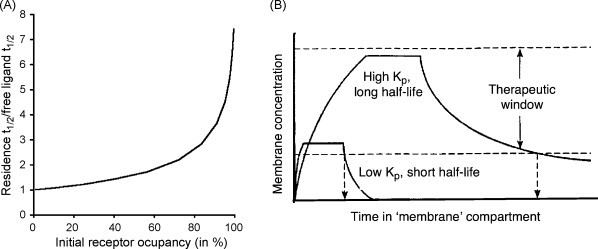
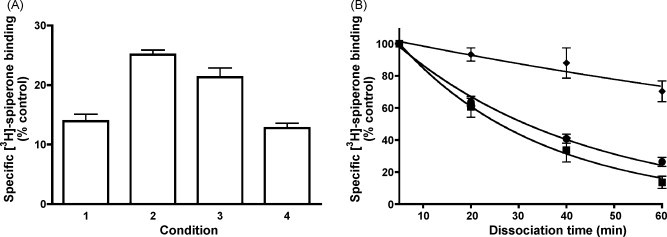
Ligand-receptor interactions are customarily described by equations that apply to solutes. Yet, most receptors are present in cell membranes so that sufficiently lipophilic ligands could reach the receptor by a two-dimensional approach within the membrane. As summarized in this review, this may affect the ligand-receptor interaction in many ways. Biophysicians calculated that, compared to a three-dimensional approach from the liquid phase, such approach could alter the time the ligands need to find a receptor. Biochemists found that ligand incorporation in lipid bilayers modifies their conformation. This, along with the depth at which the ligands reside in the bilayer, will affect the probability of successful receptor interaction. Novel mechanisms were also introduced, including "exosite" binding and ligand translocation between the receptor's alpha-helical transmembrane domains. Pharmacologists focused attention at ligand concentrations in membrane, their adsorption and release rates and the effects thereof on ligand potency and residence time at the receptor.
Figures
Similar articles
-
Does the Lipid Bilayer Orchestrate Access and Binding of Ligands to Transmembrane Orthosteric/Allosteric Sites of G Protein-Coupled Receptors?Mol Pharmacol. 2019 Nov;96(5):527-541. doi: 10.1124/mol.118.115113. Epub 2019 Apr 8. Mol Pharmacol. 2019. PMID: 30967440 Free PMC article. Review.
-
Membrane Phospholipid Analogues as Molecular Rulers to Probe the Position of the Hydrophobic Contact Point of Lysophospholipid Ligands on the Surface of G-Protein-Coupled Receptor during Membrane Approach.Biochemistry. 2020 Mar 24;59(11):1173-1201. doi: 10.1021/acs.biochem.0c00061. Epub 2020 Mar 10. Biochemistry. 2020. PMID: 32124599
-
Lipid membrane-induced optimization for ligand-receptor docking: recent tools and insights for the "membrane catalysis" model.Eur Biophys J. 2006 Jan;35(2):92-103. doi: 10.1007/s00249-005-0007-9. Epub 2005 Oct 11. Eur Biophys J. 2006. PMID: 16217647 Review.
-
Nanodiscs for immobilization of lipid bilayers and membrane receptors: kinetic analysis of cholera toxin binding to a glycolipid receptor.Anal Chem. 2008 Aug 15;80(16):6245-52. doi: 10.1021/ac8000644. Epub 2008 Jul 11. Anal Chem. 2008. PMID: 18616345
-
Phospholipid/alkanethiol bilayers for cell-surface receptor studies by surface plasmon resonance.Anal Biochem. 1995 Apr 10;226(2):342-8. doi: 10.1006/abio.1995.1234. Anal Biochem. 1995. PMID: 7793636
Cited by
-
Membrane-Facilitated Receptor Access and Binding Mechanisms of Long-Acting β2-Adrenergic Receptor Agonists.Mol Pharmacol. 2021 Oct;100(4):406-427. doi: 10.1124/molpharm.121.000285. Epub 2021 Aug 1. Mol Pharmacol. 2021. PMID: 34334369 Free PMC article.
-
The two sides of a lipid-protein story.Biophys Rev. 2016 Jun;8(2):179-191. doi: 10.1007/s12551-016-0199-5. Epub 2016 Apr 30. Biophys Rev. 2016. PMID: 28510056 Free PMC article. Review.
-
Cell membranes… and how long drugs may exert beneficial pharmacological activity in vivo.Br J Clin Pharmacol. 2016 Sep;82(3):673-82. doi: 10.1111/bcp.12996. Epub 2016 May 29. Br J Clin Pharmacol. 2016. PMID: 27135195 Free PMC article. Review.
-
Exploring Hypertension: The Role of AT1 Receptors, Sartans, and Lipid Bilayers.ACS Omega. 2024 Nov 1;9(45):44876-44890. doi: 10.1021/acsomega.4c06351. eCollection 2024 Nov 12. ACS Omega. 2024. PMID: 39554401 Free PMC article. Review.
-
Cannabinoid Receptor 2 Signalling Bias Elicited by 2,4,6-Trisubstituted 1,3,5-Triazines.Front Pharmacol. 2018 Nov 20;9:1202. doi: 10.3389/fphar.2018.01202. eCollection 2018. Front Pharmacol. 2018. PMID: 30524271 Free PMC article.
References
-
- Adam G., Delbrück M. Reduction of dimensionality in biological diffusion processes. In: Rich A., Davidson N., Freeman W.H., editors. Structural Chemistry and Molecular Biology. W.H. Freeman & Co.; San Francisco: 1968. pp. 198–215.
-
- Aiello M., Moran O., Pisciotta M., Gambale F. Interaction between dihydropyridines and phospholipid bilayers: a molecular dynamics simulation. Eur. Biophys. J. 1998;27:211–218. - PubMed
-
- Anderson G.P. In: Anderson G.P., Chapman I.D., Morley J., editors. vol. 34. Birkhauser Verlag; 1991. pp. 97–115. (In New Drugs for Asthma Therapy. Agents and Actions Supplement).
-
- Anderson G.P. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. - PubMed
-
- Anderson G.P., Lindén A., Rabe K.F. Why are long-acting beta-adrenoceptor agonists long-acting? Eur. Respir. J. 1994;7:569–578. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
