

A probabilistic approach for SNP discovery in high-throughput human resequencing data
- PMID: 19605794
- PMCID: PMC2752119
- DOI: 10.1101/gr.092072.109
A probabilistic approach for SNP discovery in high-throughput human resequencing data
Abstract
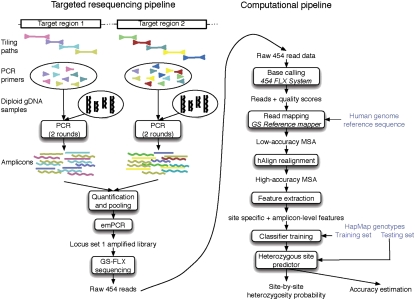
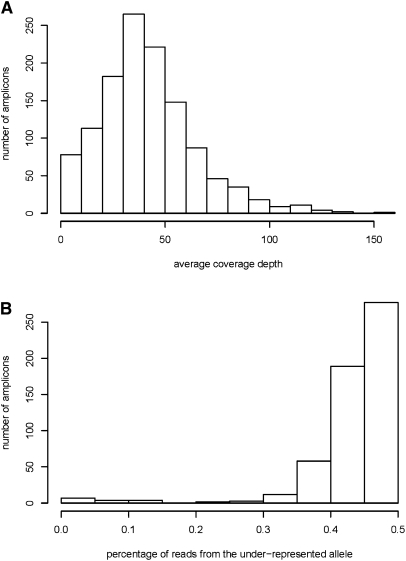
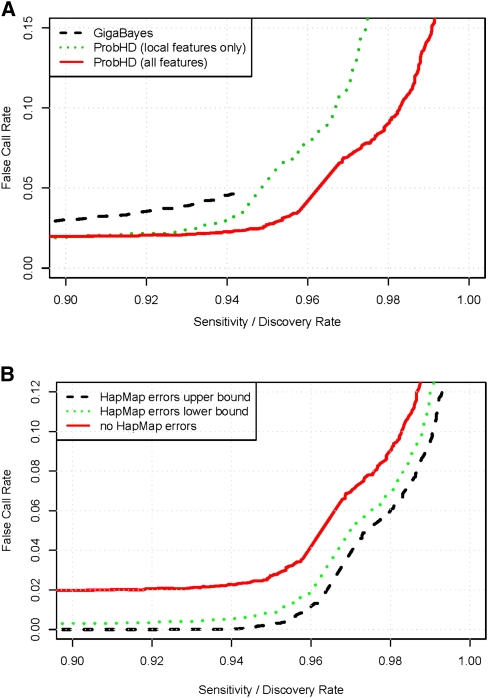
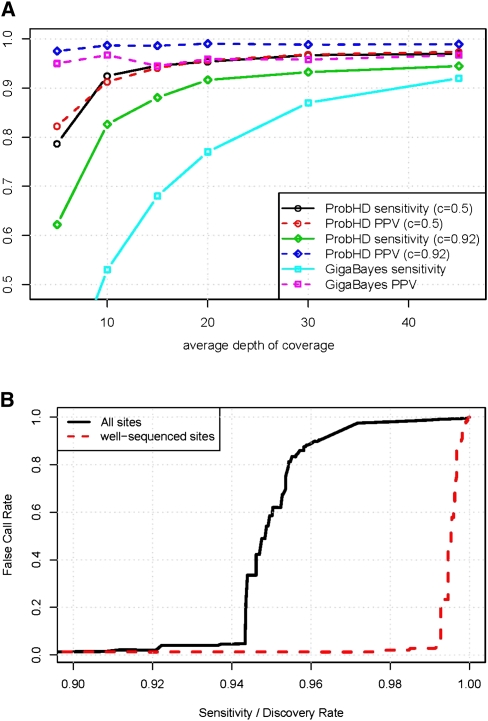
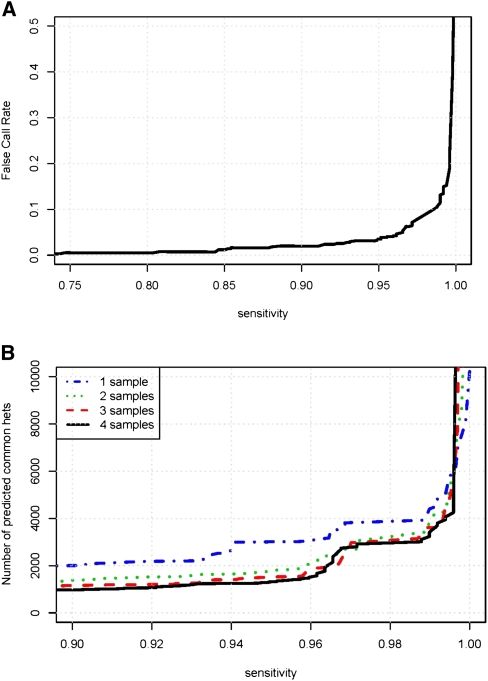
New high-throughput sequencing technologies are generating large amounts of sequence data, allowing the development of targeted large-scale resequencing studies. For these studies, accurate identification of polymorphic sites is crucial. Heterozygous sites are particularly difficult to identify, especially in regions of low coverage. We present a new strategy for identifying heterozygous sites in a single individual by using a machine learning approach that generates a heterozygosity score for each chromosomal position. Our approach also facilitates the identification of regions with unequal representation of two alleles and other poorly sequenced regions. The availability of confidence scores allows for a principled combination of sequencing results from multiple samples. We evaluate our method on a gold standard data genotype set from HapMap. We are able to classify sites in this data set as heterozygous or homozygous with 98.5% accuracy. In de novo data our probabilistic heterozygote detection ("ProbHD") is able to identify 93% of heterozygous sites at a <5% false call rate (FCR) as estimated based on independent genotyping results. In direct comparison of ProbHD with high-coverage 1000 Genomes sequencing available for a subset of our data, we observe >99.9% overall agreement for genotype calls and close to 90% agreement for heterozygote calls. Overall, our data indicate that high-throughput resequencing of human genomic regions requires careful attention to systematic biases in sample preparation as well as sequence contexts, and that their impact can be alleviated by machine learning-based sequence analyses allowing more accurate extraction of true DNA variants.
Figures
Similar articles
-
SNP detection for massively parallel whole-genome resequencing.Genome Res. 2009 Jun;19(6):1124-32. doi: 10.1101/gr.088013.108. Epub 2009 May 6. Genome Res. 2009. PMID: 19420381 Free PMC article.
-
Accurate detection and genotyping of SNPs utilizing population sequencing data.Genome Res. 2010 Apr;20(4):537-45. doi: 10.1101/gr.100040.109. Epub 2010 Feb 11. Genome Res. 2010. PMID: 20150320 Free PMC article.
-
Genome-wide analysis of allelic expression imbalance in human primary cells by high-throughput transcriptome resequencing.Hum Mol Genet. 2010 Jan 1;19(1):122-34. doi: 10.1093/hmg/ddp473. Hum Mol Genet. 2010. PMID: 19825846 Free PMC article.
-
Genome Wide Sampling Sequencing for SNP Genotyping: Methods, Challenges and Future Development.Int J Biol Sci. 2016 Jan 1;12(1):100-8. doi: 10.7150/ijbs.13498. eCollection 2016. Int J Biol Sci. 2016. PMID: 26722221 Free PMC article. Review.
-
Enabling large-scale pharmacogenetic studies by high-throughput mutation detection and genotyping technologies.Clin Chem. 2001 Feb;47(2):164-72. Clin Chem. 2001. PMID: 11159763 Review.
Cited by
-
A cross-sample statistical model for SNP detection in short-read sequencing data.Nucleic Acids Res. 2012 Jan;40(1):e5. doi: 10.1093/nar/gkr851. Epub 2011 Nov 7. Nucleic Acids Res. 2012. PMID: 22064853 Free PMC article.
-
Next generation sequence analysis and computational genomics using graphical pipeline workflows.Genes (Basel). 2012 Aug 30;3(3):545-75. doi: 10.3390/genes3030545. Genes (Basel). 2012. PMID: 23139896 Free PMC article.
-
Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population.Science. 2009 Dec 18;326(5960):1663-7. doi: 10.1126/science.1178357. Science. 2009. PMID: 20019281 Free PMC article.
-
Bioinformatics for next generation sequencing data.Genes (Basel). 2010 Sep 14;1(2):294-307. doi: 10.3390/genes1020294. Genes (Basel). 2010. PMID: 24710047 Free PMC article.
-
Inferring demography from runs of homozygosity in whole-genome sequence, with correction for sequence errors.Mol Biol Evol. 2013 Sep;30(9):2209-23. doi: 10.1093/molbev/mst125. Epub 2013 Jul 10. Mol Biol Evol. 2013. PMID: 23842528 Free PMC article.
References
-
- Albert TJ, Molla MN, Muzny DM, Nazareth L, Wheeler D, Song X, Richmond TA, Middle CM, Rodesch MJ, Packard CJ, et al. Direct selection of human genomic loci by microarray hybridization. Nat Methods. 2007;4:903–905. - PubMed
-
- Breiman L. Random forests. Mach Learn. 2001;45:5–32.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous