

Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?
- PMID: 19584893
- PMCID: PMC2853768
- DOI: 10.1038/nrn2670
Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick?
Abstract
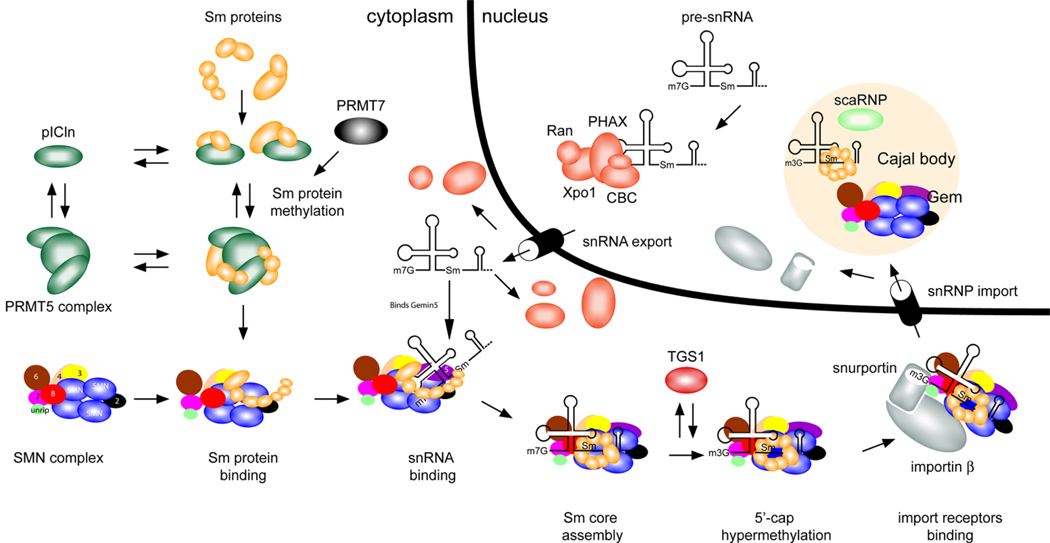
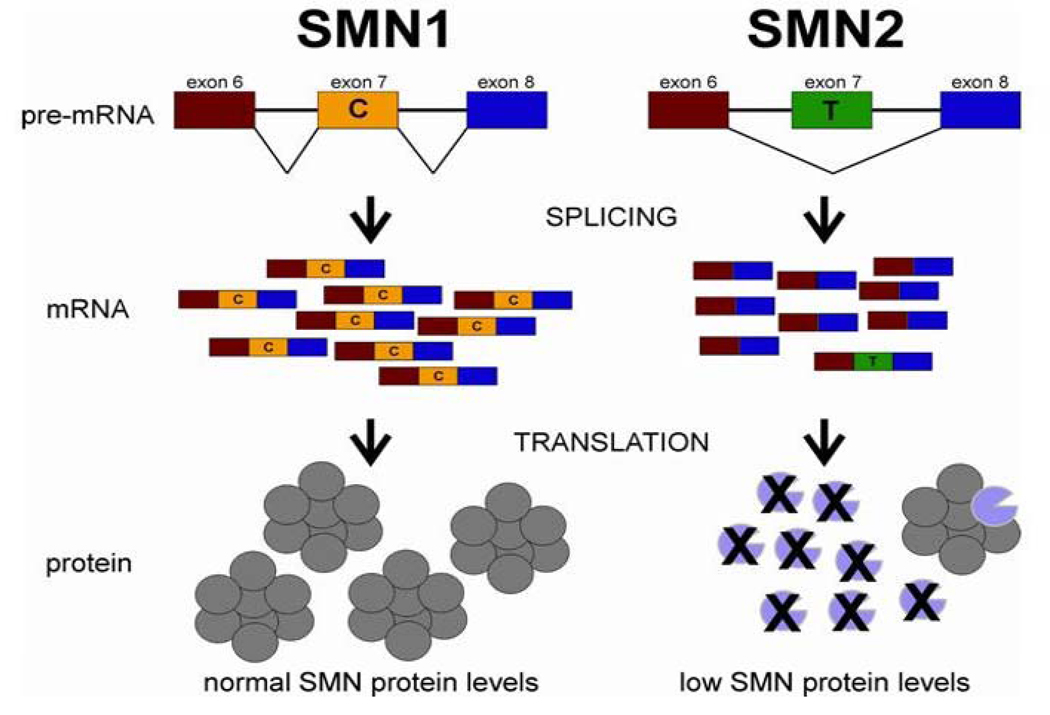
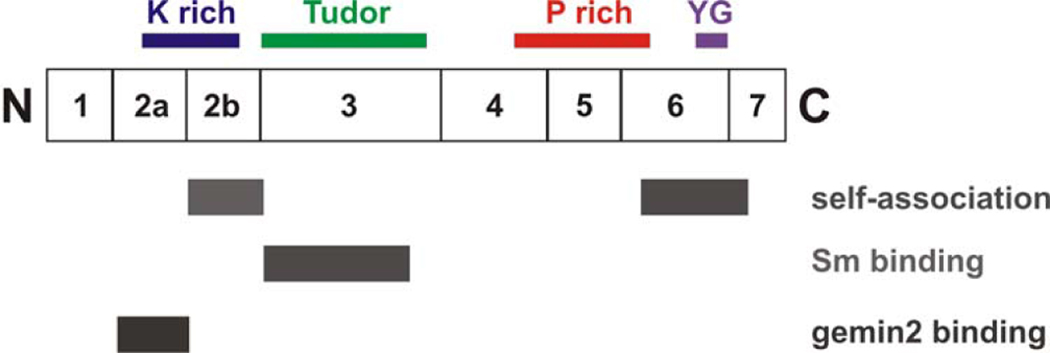
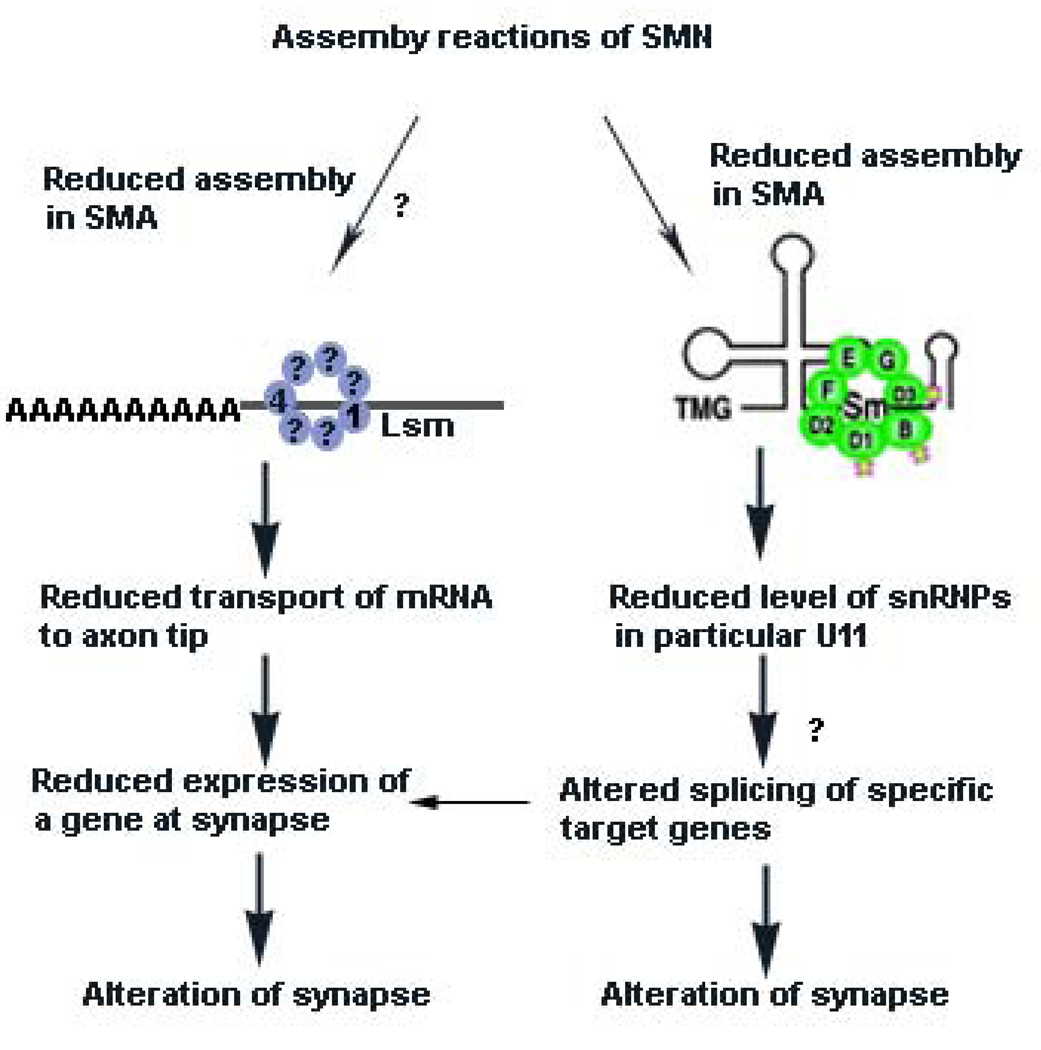
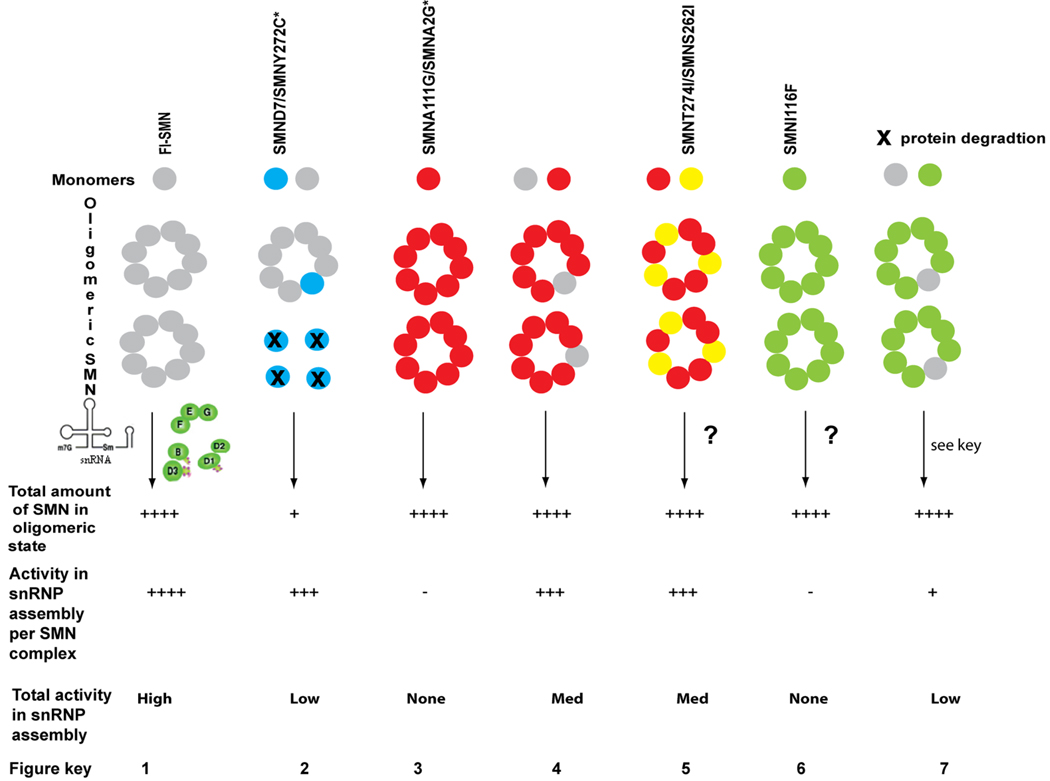
Many neurogenetic disorders are caused by the mutation of ubiquitously expressed genes. One such disorder, spinal muscular atrophy, is caused by loss or mutation of the survival motor neuron1 gene (SMN1), leading to reduced SMN protein levels and a selective dysfunction of motor neurons. SMN, together with partner proteins, functions in the assembly of small nuclear ribonucleoproteins (snRNPs), which are important for pre-mRNA splicing. It has also been suggested that SMN might function in the assembly of other ribonucleoprotein complexes. Two hypotheses have been proposed to explain the molecular dysfunction that gives rise to spinal muscular atrophy (SMA) and its specificity to a particular group of neurons. The first hypothesis states that the loss of SMN's well-known function in snRNP assembly causes an alteration in the splicing of a specific gene (or genes). The second hypothesis proposes that SMN is crucial for the transport of mRNA in neurons and that disruption of this function results in SMA.
Figures
Similar articles
-
Spinal muscular atrophy: Selective motor neuron loss and global defect in the assembly of ribonucleoproteins.Brain Res. 2018 Aug 15;1693(Pt A):92-97. doi: 10.1016/j.brainres.2018.02.022. Epub 2018 Feb 17. Brain Res. 2018. PMID: 29462610 Review.
-
RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns.Nucleic Acids Res. 2017 Jan 9;45(1):395-416. doi: 10.1093/nar/gkw731. Epub 2016 Aug 23. Nucleic Acids Res. 2017. PMID: 27557711 Free PMC article.
-
What Genetics Has Told Us and How It Can Inform Future Experiments for Spinal Muscular Atrophy, a Perspective.Int J Mol Sci. 2021 Aug 6;22(16):8494. doi: 10.3390/ijms22168494. Int J Mol Sci. 2021. PMID: 34445199 Free PMC article.
-
Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs.PLoS One. 2007 Sep 26;2(9):e921. doi: 10.1371/journal.pone.0000921. PLoS One. 2007. PMID: 17895963 Free PMC article.
-
Spliceosomal small nuclear ribonucleoprotein biogenesis defects and motor neuron selectivity in spinal muscular atrophy.Brain Res. 2012 Jun 26;1462:93-9. doi: 10.1016/j.brainres.2012.02.051. Epub 2012 Feb 28. Brain Res. 2012. PMID: 22424789 Free PMC article. Review.
Cited by
-
Regulatory roles of RNA binding proteins in the nervous system of C. elegans.Front Mol Neurosci. 2015 Jan 12;7:100. doi: 10.3389/fnmol.2014.00100. eCollection 2014. Front Mol Neurosci. 2015. PMID: 25628531 Free PMC article. Review.
-
Genetic Interactions between the Members of the SMN-Gemins Complex in Drosophila.PLoS One. 2015 Jun 22;10(6):e0130974. doi: 10.1371/journal.pone.0130974. eCollection 2015. PLoS One. 2015. PMID: 26098872 Free PMC article.
-
The SMN-ribosome interplay: a new opportunity for Spinal Muscular Atrophy therapies.Biochem Soc Trans. 2024 Feb 28;52(1):465-479. doi: 10.1042/BST20231116. Biochem Soc Trans. 2024. PMID: 38391004 Free PMC article. Review.
-
TSUNAMI: an antisense method to phenocopy splicing-associated diseases in animals.Genes Dev. 2012 Aug 15;26(16):1874-84. doi: 10.1101/gad.197418.112. Genes Dev. 2012. PMID: 22895255 Free PMC article.
-
A link between agrin signalling and Cav3.2 at the neuromuscular junction in spinal muscular atrophy.Sci Rep. 2022 Nov 8;12(1):18960. doi: 10.1038/s41598-022-23703-x. Sci Rep. 2022. PMID: 36347955 Free PMC article.
References
-
- Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. - PubMed
-
- Melki J. Spinal muscular atrophy. Curr Opin Neurol. 1997;10:381–385. - PubMed
-
-
McAndrew PE, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60:1411–1422.First copy analysis of
SMN1 andSMN2 and clear indication that copy number of SMN2 modifies SMA phenotype
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
