

Review
doi: 10.1146/annurev.biochem.77.061906.092014.
Super-resolution fluorescence microscopy
Affiliations
- PMID: 19489737
- PMCID: PMC2835776
- DOI: 10.1146/annurev.biochem.77.061906.092014
Item in Clipboard
Review
Super-resolution fluorescence microscopy
Annu Rev Biochem.
2009.
Abstract
Achieving a spatial resolution that is not limited by the diffraction of light, recent developments of super-resolution fluorescence microscopy techniques allow the observation of many biological structures not resolvable in conventional fluorescence microscopy. New advances in these techniques now give them the ability to image three-dimensional (3D) structures, measure interactions by multicolor colocalization, and record dynamic processes in living cells at the nanometer scale. It is anticipated that super-resolution fluorescence microscopy will become a widely used tool for cell and tissue imaging to provide previously unobserved details of biological structures and processes.
Figures

The PSF of a common oil immersion objective with NA = 1.40, showing the focal spot of 550 nm light in a medium with refractive index n = 1.515. The intensity distribution in the x-z plane of the focus spot is computed numerically and shown in the upper panel and the FWHM in the lateral and axial directions are 220 nm and 520 nm, respectively.

The principle of STED microscopy. (a) The process of stimulated emission. A ground state (S0) fluorophore can absorb a photon from the excitation light and jump to the excited state (S1). Spontaneous fluorescence emissionbrings the fluorophore back to the ground state. Stimulated emission happens when the excited state fluorophore encounters another photon with wavelength comparable to the energy difference between the ground and excited state. (b) Schematic drawing of a STED microscope. The excitation laser and STED laser are combined and focused into the sample through the objective. A phase mask is placed in the light path of the STED laser to create a specific pattern at the objective focal point. (c) In the xy mode, a donut-shaped STED laser is applied with the zero point overlapped with the maximum of the excitation laser focus. With saturated depletion, fluorescence from regions near the zero point is suppressed, leading to a decreased size of the effective PSF.
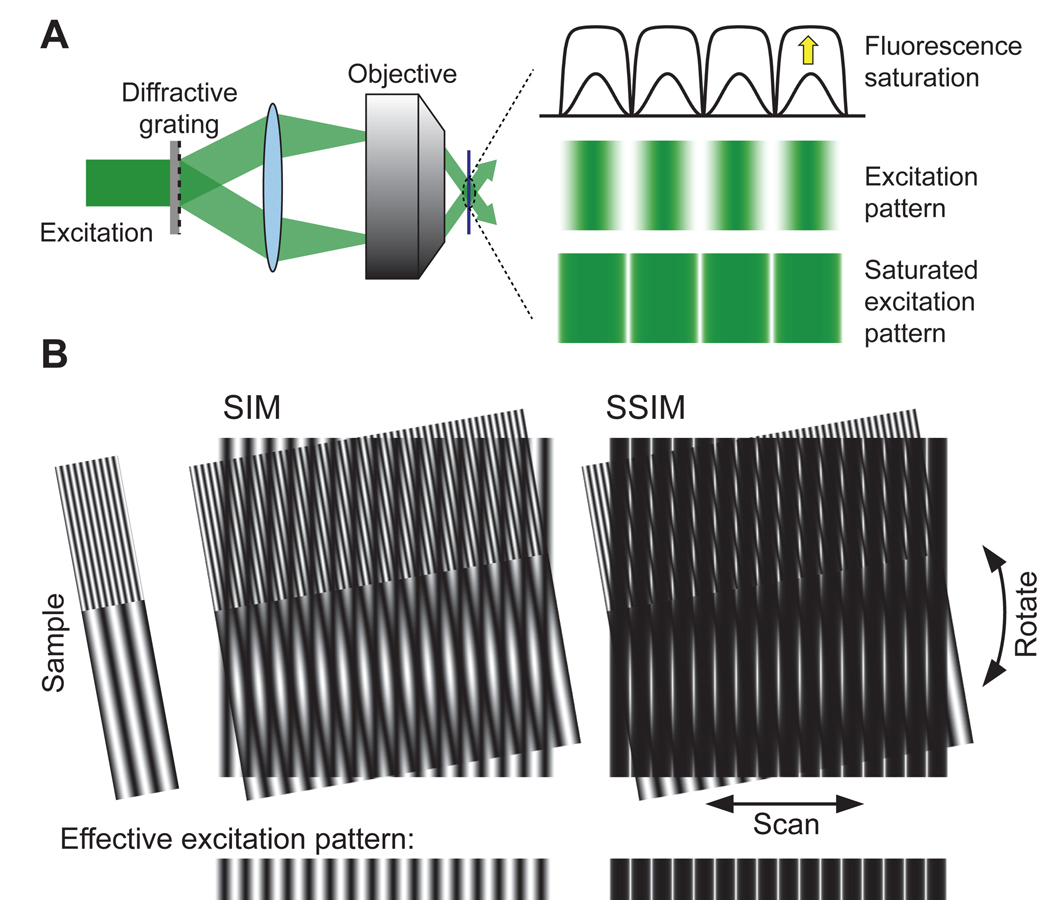
The principle of SSIM. (a) The generation of the illumination pattern. A diffractive grating in the excitation path splits the light into two beams. Their interference after emerging from the objective and reaching the sample creates a sinusoidal illumination pattern with alternating peaks and zero points. Strong excitation light saturates the fluorescence emission at the peaks without exciting fluorophores at the zero points, leading to sharp dark regions in the effective illumination pattern. (b) Resolving fine structures with SIM and SSIM. When apply a sinusoidal illumination pattern to a sample, moiré pattern at a significantly lower spatial frequency than that of the sample can be generated and imaged by the microscope (SIM panel, lower part). Multiple images resulted from scanning and rotating the excitation pattern are then used to reconstruct the sample structure. However, fine structures with spatial frequencies much higher than the illumination pattern are still indiscernible, as mixing the two does not generate significantly lower spatial frequency in the moiré pattern (SIM panel, upper part). SSIM introduces high frequency component into the illumination pattern, allowing features far below the diffraction limit to be resolved (as is evident by the appearance of low frequency moiré pattern in the upper part of the SSIM panel).
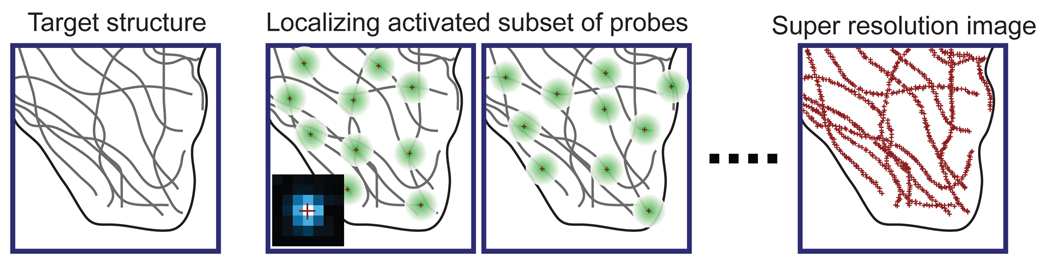
The principle of stochastic optical reconstruction microscopy (STORM), photoactivated localization microscopy (PALM), and fluorescence photoactivation localization microscopy (FPALM). Different fluorescent probes marking the sample structure are activated at different time points, allowing subsets of fluorophores to be imaged without spatial overlap and to be localized to high precision. Iterating the activation and imaging process allows the position of many fluorescent probes to be determined and a super-resolution image is then reconstructed from the positions of a large number of localized probe molecules. The lower left inset of the second panel shows an experimental image of a single fluorescent dye (blue) and the high-precision localization of the molecule (red cross).
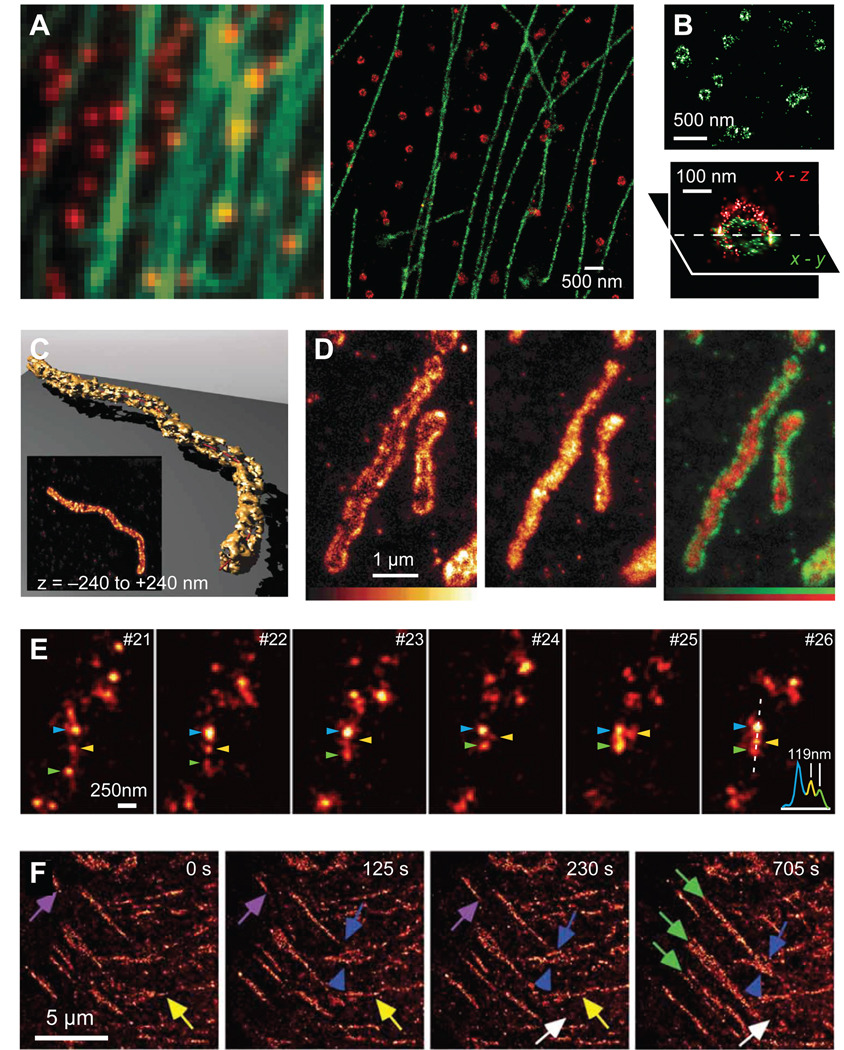
Examples of super-resolution images of biological samples. (a) Two-color STORM imaging of immunostained microtubule (green) and clathrin-coated pits (red) (From Reference . Reprinted with permission from AAAS). Microtubules overlapped in the conventional fluorescence image (left panel) are resolved in the super-resolution image (right panel). Clathrin-coated pits and microtubules with substantial colocalization in the conventional fluorescence image are clearly separated. (b) 3D STORM images of clathrin-coated pits (From Reference 52). Reprinted with permission from AAAS). Although shown as filled circles in the 2D STORM image (left panel), the 100-nm thick x-y section across the rim of two clathrin-coated pits (center panel), and the 3D perspective of a 50-nm thick x-y and a x-z section of one pit (right panel) reveal their hemispherical cage shape. (c) 3D two-color STED microscopy of the mitochondria outer membrane immunostained for the tom20 protein (left panel) and the mitochondria matrix immunostained for the matrix protein Hsp60 (center panel) (Reprinted by permission from Macmillan Publishers Ltd.: Nature Methods, Reference , copyright 2008). In the two-color overlaid image of the 45-nm thick x-y section (right panel), the outer membrane tom20 proteins (green) are shown to enclose Hsp60 (red). (d) STED images of synaptic vesicles in live neurons at a frame rate of 28 fps (From Reference . Reprinted with permission from AAAS). A spatial resolution of ~60 nm allows individual immunostained synaptic vesicles (arrow heads) to be resolved and tracked. (e) Live-cell imaging of a focal adhesion complex (Reprinted by permission from Macmillan Publishers Ltd.: Nature Methods, Reference , copyright 2008). Super-resolution PALM images of EosFP-tagged paxillin are reconstructed at 60-s interval. Focal adhesion complexes with different modes of movement are marked with colored arrows.
Similar articles
-
3D super-resolution imaging by localization microscopy.Methods Mol Biol. 2015;1232:123-36. doi: 10.1007/978-1-4939-1752-5_11. Methods Mol Biol. 2015. PMID: 25331133
-
Visualizing and discovering cellular structures with super-resolution microscopy.Science. 2018 Aug 31;361(6405):880-887. doi: 10.1126/science.aau1044. Epub 2018 Aug 30. Science. 2018. PMID: 30166485 Free PMC article. Review.
-
High-speed super-resolution imaging of rotationally symmetric structures using SPEED microscopy and 2D-to-3D transformation.Nat Protoc. 2021 Jan;16(1):532-560. doi: 10.1038/s41596-020-00440-x. Epub 2020 Dec 14. Nat Protoc. 2021. PMID: 33318694 Free PMC article.
-
Recent advances in super-resolution fluorescence imaging and its applications in biology.J Genet Genomics. 2013 Dec 20;40(12):583-95. doi: 10.1016/j.jgg.2013.11.003. Epub 2013 Nov 23. J Genet Genomics. 2013. PMID: 24377865 Review.
-
Breaking the diffraction barrier: super-resolution imaging of cells.Cell. 2010 Dec 23;143(7):1047-58. doi: 10.1016/j.cell.2010.12.002. Cell. 2010. PMID: 21168201 Free PMC article.
Cited by
-
Holotomography: Refractive Index as an Intrinsic Imaging Contrast for 3-D Label-Free Live Cell Imaging.Adv Exp Med Biol. 2021;1310:211-238. doi: 10.1007/978-981-33-6064-8_10. Adv Exp Med Biol. 2021. PMID: 33834439
-
Subdiffraction-resolution fluorescence microscopy reveals a domain of the centrosome critical for pericentriolar material organization.Nat Cell Biol. 2012 Nov;14(11):1159-68. doi: 10.1038/ncb2597. Epub 2012 Oct 21. Nat Cell Biol. 2012. PMID: 23086239 Free PMC article.
-
Multiscale perspectives of virus entry via endocytosis.Virol J. 2013 Jun 5;10:177. doi: 10.1186/1743-422X-10-177. Virol J. 2013. PMID: 23734580 Free PMC article. Review.
-
Quantifying the minimum localization uncertainty of image scanning localization microscopy.Biophys Rep (N Y). 2024 Jan 20;4(1):100143. doi: 10.1016/j.bpr.2024.100143. eCollection 2024 Mar 13. Biophys Rep (N Y). 2024. PMID: 38380223 Free PMC article.
-
Ultrasensitive and long-range transverse displacement metrology with polarization-encoded metasurface.Sci Adv. 2022 Oct 14;8(41):eadd1973. doi: 10.1126/sciadv.add1973. Epub 2022 Oct 12. Sci Adv. 2022. PMID: 36223465 Free PMC article.
References
-
- Hell SW, Wichmann J. Breaking the diffraction resolution limit by stimulated-emission: stimulated emission- depletion fluorescence microscopy. Opt. Lett. 1994;19:780–782. - PubMed
-
- Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999;24:954–956. - PubMed
-
- Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
