

The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers
- PMID: 19478061
- PMCID: PMC2687998
- DOI: 10.1073/pnas.0900571106
The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers
Abstract
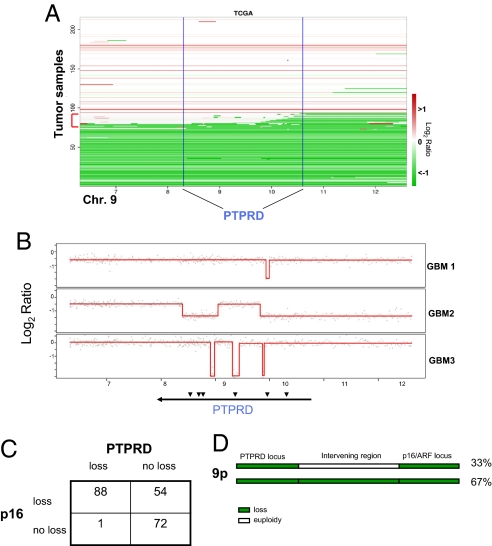
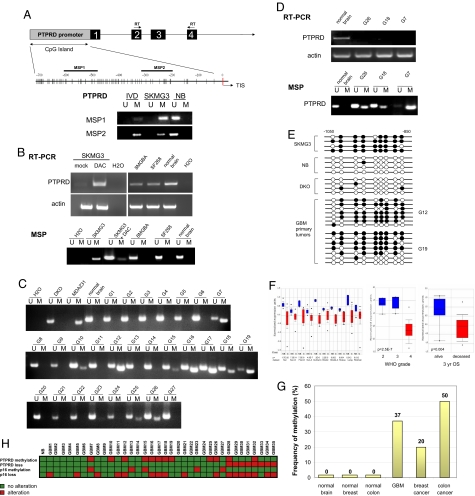
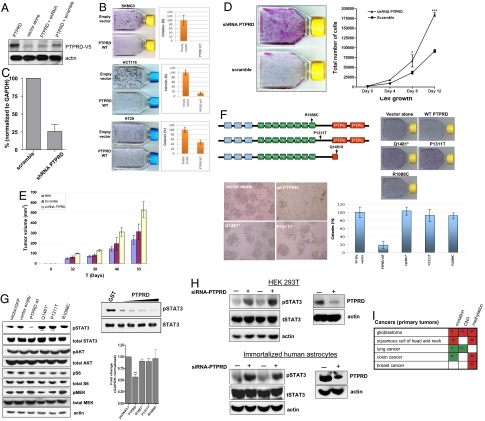
Tyrosine phosphorylation plays a critical role in regulating cellular function and is a central feature in signaling cascades involved in oncogenesis. The regulation of tyrosine phosphorylation is coordinately controlled by kinases and phosphatases (PTPs). Whereas activation of tyrosine kinases has been shown to play vital roles in tumor development, the role of PTPs is much less well defined. Here, we show that the receptor protein tyrosine phosphatase delta (PTPRD) is frequently inactivated in glioblastoma multiforme (GBM), a deadly primary neoplasm of the brain. PTPRD is a target of deletion in GBM, often via focal intragenic loss. In GBM tumors that do not possess deletions in PTPRD, the gene is frequently subject to cancer-specific epigenetic silencing via promoter CpG island hypermethylation (37%). Sequencing of the PTPRD gene in GBM and other primary human tumors revealed that the gene is mutated in 6% of GBMs, 13% of head and neck squamous cell carcinomas, and in 9% of lung cancers. These mutations were deleterious. In total, PTPRD inactivation occurs in >50% of GBM tumors, and loss of expression predicts for poor prognosis in glioma patients. Wild-type PTPRD inhibits the growth of GBM and other tumor cells, an effect not observed with PTPRD alleles harboring cancer-specific mutations. Human astrocytes lacking PTPRD exhibited increased growth. PTPRD was found to dephosphorylate the oncoprotein STAT3. These results implicate PTPRD as a tumor suppressor on chromosome 9p that is involved in the development of GBMs and multiple human cancers.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Loss of the tyrosine phosphatase PTPRD leads to aberrant STAT3 activation and promotes gliomagenesis.Proc Natl Acad Sci U S A. 2014 Jun 3;111(22):8149-54. doi: 10.1073/pnas.1401952111. Epub 2014 May 19. Proc Natl Acad Sci U S A. 2014. PMID: 24843164 Free PMC article.
-
Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma.Cancer Res. 2008 Dec 15;68(24):10300-6. doi: 10.1158/0008-5472.CAN-08-3272. Cancer Res. 2008. PMID: 19074898 Free PMC article.
-
Recurrent epigenetic silencing of the PTPRD tumor suppressor in laryngeal squamous cell carcinoma.Tumour Biol. 2017 Mar;39(3):1010428317691427. doi: 10.1177/1010428317691427. Tumour Biol. 2017. PMID: 28345455
-
Tyrosine phosphatases as a superfamily of tumor suppressors in colorectal cancer.Acta Biochim Pol. 2011;58(4):467-70. Epub 2011 Dec 6. Acta Biochim Pol. 2011. PMID: 22146137 Review.
-
Protein Tyrosine Phosphatases as Potential Regulators of STAT3 Signaling.Int J Mol Sci. 2018 Sep 11;19(9):2708. doi: 10.3390/ijms19092708. Int J Mol Sci. 2018. PMID: 30208623 Free PMC article. Review.
Cited by
-
Genomic Alteration in Head and Neck Squamous Cell Carcinoma (HNSCC) Cell Lines Inferred from Karyotyping, Molecular Cytogenetics, and Array Comparative Genomic Hybridization.PLoS One. 2016 Aug 8;11(8):e0160901. doi: 10.1371/journal.pone.0160901. eCollection 2016. PLoS One. 2016. PMID: 27501229 Free PMC article.
-
Gene deletions and amplifications in human hepatocellular carcinomas: correlation with hepatocyte growth regulation.Am J Pathol. 2012 Apr;180(4):1495-508. doi: 10.1016/j.ajpath.2011.12.021. Epub 2012 Feb 8. Am J Pathol. 2012. PMID: 22326833 Free PMC article.
-
Viral manipulation of STAT3: Evade, exploit, and injure.PLoS Pathog. 2018 Mar 15;14(3):e1006839. doi: 10.1371/journal.ppat.1006839. eCollection 2018 Mar. PLoS Pathog. 2018. PMID: 29543893 Free PMC article. Review.
-
Mapping the protein-protein and genetic interactions of cancer to guide precision medicine.Curr Opin Genet Dev. 2019 Feb;54:110-117. doi: 10.1016/j.gde.2019.04.005. Epub 2019 Jul 6. Curr Opin Genet Dev. 2019. PMID: 31288129 Free PMC article. Review.
-
Cross-talk between phospho-STAT3 and PLCγ1 plays a critical role in colorectal tumorigenesis.Mol Cancer Res. 2011 Oct;9(10):1418-28. doi: 10.1158/1541-7786.MCR-11-0147. Epub 2011 Aug 12. Mol Cancer Res. 2011. PMID: 21840932 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
