

Disentangling molecular relationships with a causal inference test
- PMID: 19473544
- PMCID: PMC3224661
- DOI: 10.1186/1471-2156-10-23
Disentangling molecular relationships with a causal inference test
Abstract
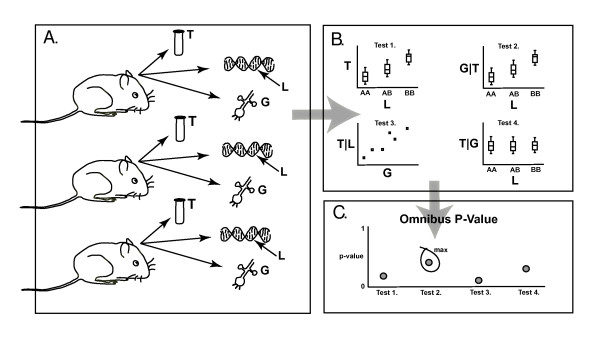
Background: There has been intense effort over the past couple of decades to identify loci underlying quantitative traits as a key step in the process of elucidating the etiology of complex diseases. Recently there has been some effort to coalesce non-biased high-throughput data, e.g. high density genotyping and genome wide RNA expression, to drive understanding of the molecular basis of disease. However, a stumbling block has been the difficult question of how to leverage this information to identify molecular mechanisms that explain quantitative trait loci (QTL). We have developed a formal statistical hypothesis test, resulting in a p-value, to quantify uncertainty in a causal inference pertaining to a measured factor, e.g. a molecular species, which potentially mediates a known causal association between a locus and a quantitative trait.
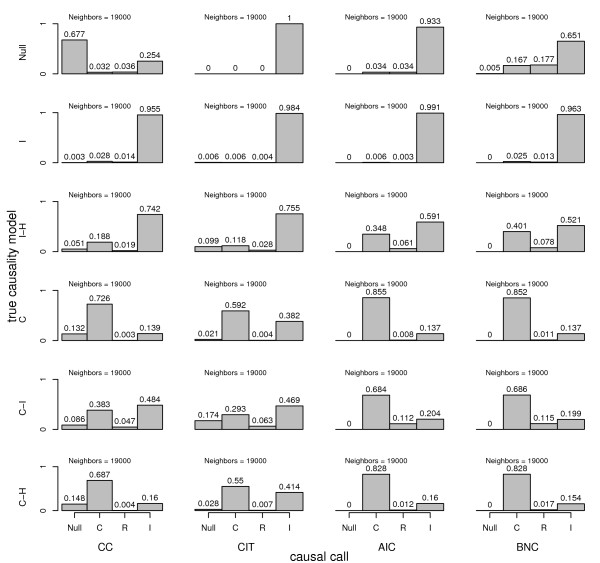
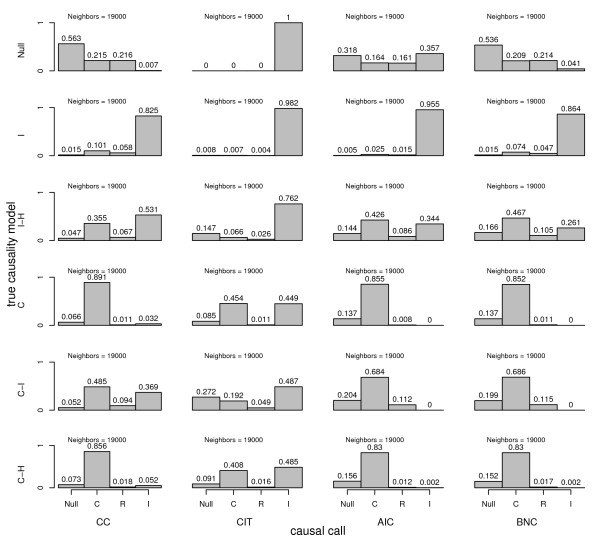
Results: We treat the causal inference as a 'chain' of mathematical conditions that must be satisfied to conclude that the potential mediator is causal for the trait, where the inference is only as good as the weakest link in the chain. P-values are computed for the component conditions, which include tests of linkage and conditional independence. The Intersection-Union Test, in which a series of statistical tests are combined to form an omnibus test, is then employed to generate the overall test result. Using computer simulated mouse crosses, we show that type I error is low under a variety of conditions that include hidden variables and reactive pathways. We show that power under a simple causal model is comparable to other model selection techniques as well as Bayesian network reconstruction methods. We further show empirically that this method compares favorably to Bayesian network reconstruction methods for reconstructing transcriptional regulatory networks in yeast, recovering 7 out of 8 experimentally validated regulators.
Conclusion: Here we propose a novel statistical framework in which existing notions of causal mediation are formalized into a hypothesis test, thus providing a standard quantitative measure of uncertainty in the form of a p-value. The method is theoretically and computationally accessible and with the provided software may prove a useful tool in disentangling molecular relationships.
Figures




Similar articles
-
Uncovering the genetic landscape for multiple sleep-wake traits.PLoS One. 2009;4(4):e5161. doi: 10.1371/journal.pone.0005161. Epub 2009 Apr 10. PLoS One. 2009. PMID: 19360106 Free PMC article.
-
Using stochastic causal trees to augment Bayesian networks for modeling eQTL datasets.BMC Bioinformatics. 2011 Jan 6;12:7. doi: 10.1186/1471-2105-12-7. BMC Bioinformatics. 2011. PMID: 21211042 Free PMC article.
-
A Bayesian framework for inference of the genotype-phenotype map for segregating populations.Genetics. 2011 Apr;187(4):1163-70. doi: 10.1534/genetics.110.123273. Epub 2011 Jan 17. Genetics. 2011. PMID: 21242536 Free PMC article.
-
Current progress on statistical methods for mapping quantitative trait loci from inbred line crosses.J Biopharm Stat. 2010 Mar;20(2):454-81. doi: 10.1080/10543400903572845. J Biopharm Stat. 2010. PMID: 20309768 Review.
-
Application of Causal Inference to Genomic Analysis: Advances in Methodology.Front Genet. 2018 Jul 10;9:238. doi: 10.3389/fgene.2018.00238. eCollection 2018. Front Genet. 2018. PMID: 30042787 Free PMC article. Review.
Cited by
-
Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders.J Am Coll Cardiol. 2015 Mar 3;65(8):830-845. doi: 10.1016/j.jacc.2014.12.033. J Am Coll Cardiol. 2015. PMID: 25720628 Free PMC article. Review.
-
Genetic identification of Ly75 as a novel quantitative trait gene for resistance to obesity in mice.Sci Rep. 2018 Dec 5;8(1):17658. doi: 10.1038/s41598-018-36073-0. Sci Rep. 2018. PMID: 30518881 Free PMC article.
-
Multiscale Analysis of Independent Alzheimer's Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus.Neuron. 2018 Jul 11;99(1):64-82.e7. doi: 10.1016/j.neuron.2018.05.023. Epub 2018 Jun 21. Neuron. 2018. PMID: 29937276 Free PMC article.
-
Multiomics Analyses Identify Proline Endopeptidase-Like Protein As a Key Regulator of Protein Trafficking, a Pathway Underlying Alzheimer's Disease Pathogenesis.Mol Pharmacol. 2023 Jul;104(1):1-16. doi: 10.1124/molpharm.122.000641. Epub 2023 May 5. Mol Pharmacol. 2023. PMID: 37147110 Free PMC article.
-
A Strategy for Identifying Quantitative Trait Genes Using Gene Expression Analysis and Causal Analysis.Genes (Basel). 2017 Nov 27;8(12):347. doi: 10.3390/genes8120347. Genes (Basel). 2017. PMID: 29186889 Free PMC article. Review.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
