

TRPM2 functions as a lysosomal Ca2+-release channel in beta cells
- PMID: 19454650
- PMCID: PMC2779714
- DOI: 10.1126/scisignal.2000278
TRPM2 functions as a lysosomal Ca2+-release channel in beta cells
Abstract
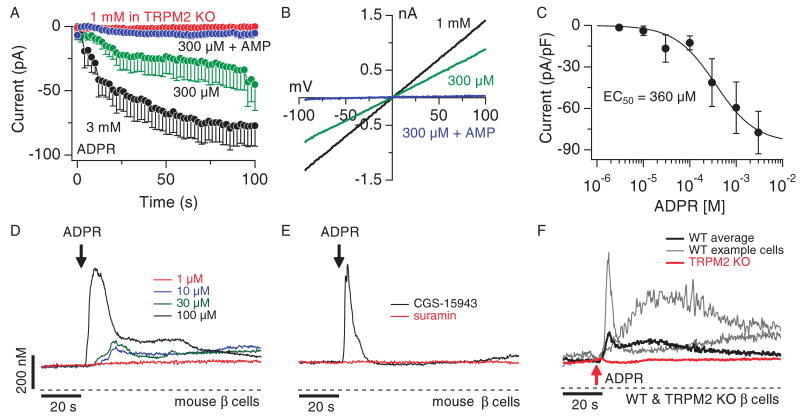
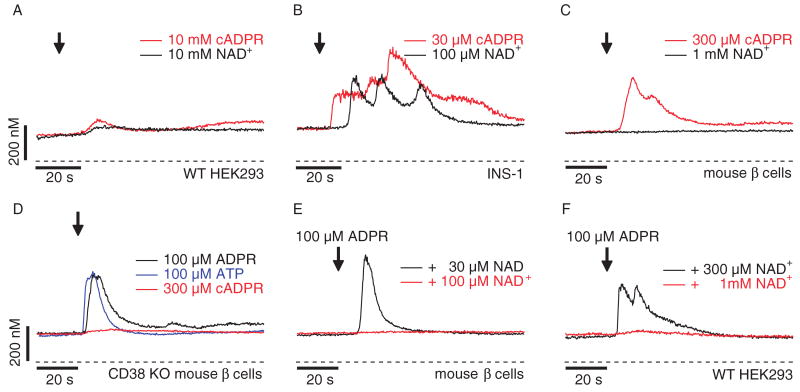
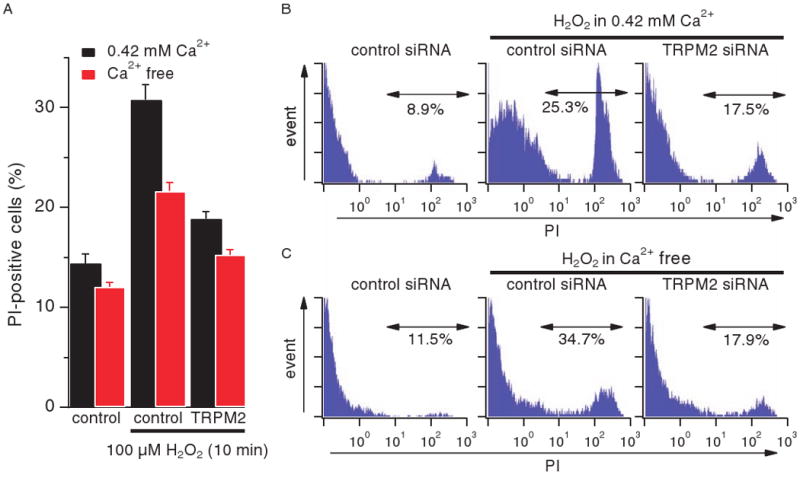
TRPM2 is a Ca2+-permeable cation channel that is specifically activated by adenosine diphosphoribose (ADPR). Channel activation in the plasma membrane leads to Ca2+ influx and has been linked to apoptotic mechanisms. The primary agonist, ADPR, is produced both extra- and intracellularly and causes increases in intracellular calcium concentration ([Ca2+]i), but the mechanisms involved are not understood. Using short interfering RNA and a knockout mouse, we report that TRPM2, in addition to its role as a plasma membrane channel, also functions as a Ca2+-release channel activated by intracellular ADPR in a lysosomal compartment. We show that both functions of TRPM2 are critically linked to hydrogen peroxide-induced beta cell death. Additionally, extracellular ADPR production by the ectoenzyme CD38 from its substrates NAD+ (nicotinamide adenine dinucleotide) or cADPR causes IP3-dependent Ca2+ release via P2Y and adenosine receptors. Thus, ADPR and TRPM2 represent multimodal signaling elements regulating Ca2+ mobilization in beta cells through membrane depolarization, Ca2+ influx, and release of Ca2+ from intracellular stores.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
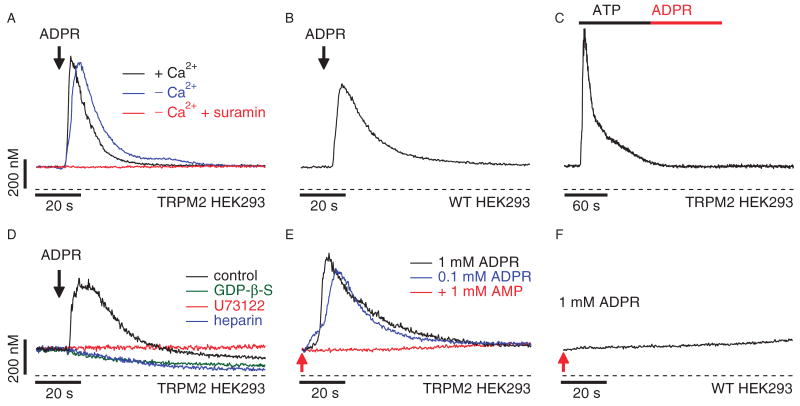
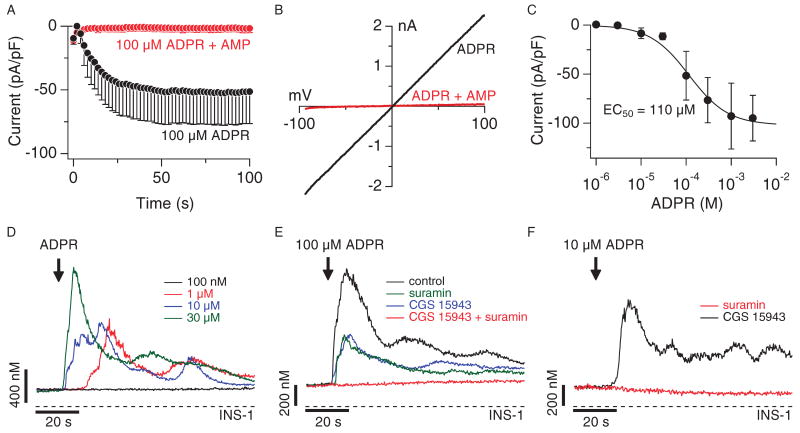
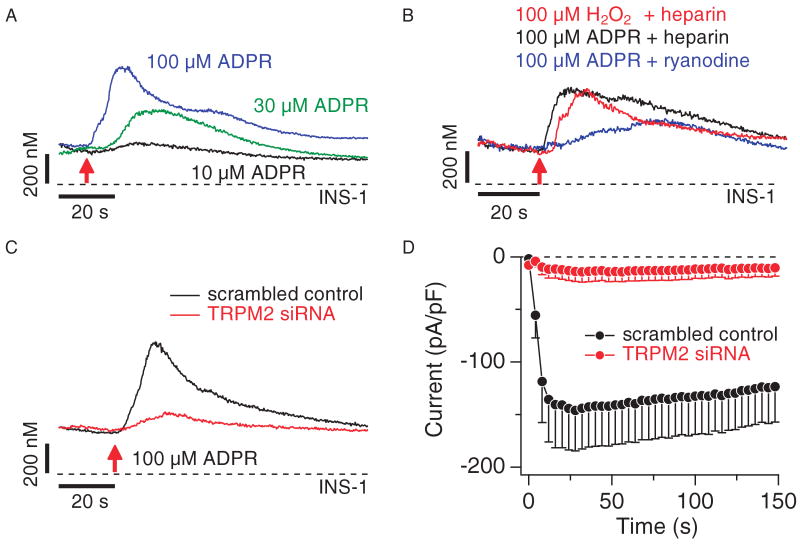
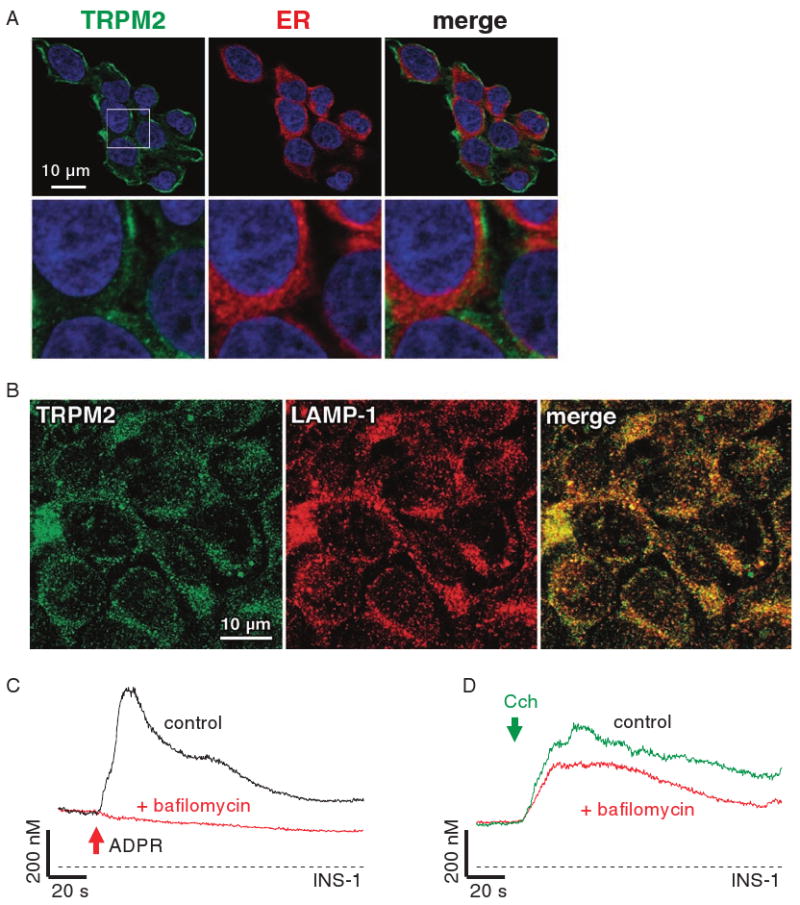
Similar articles
-
Extracellular Adenosine Diphosphate Ribose Mobilizes Intracellular Ca2+ via Purinergic-Dependent Ca2+ Pathways in Rat Pulmonary Artery Smooth Muscle Cells.Cell Physiol Biochem. 2015;37(5):2043-59. doi: 10.1159/000438564. Epub 2015 Nov 20. Cell Physiol Biochem. 2015. PMID: 26584302
-
A calcium influx pathway regulated separately by oxidative stress and ADP-Ribose in TRPM2 channels: single channel events.Neurochem Res. 2008 Jul;33(7):1256-62. doi: 10.1007/s11064-007-9577-5. Epub 2008 Feb 8. Neurochem Res. 2008. PMID: 18259858
-
Chemotaxis of mouse bone marrow neutrophils and dendritic cells is controlled by adp-ribose, the major product generated by the CD38 enzyme reaction.J Immunol. 2007 Dec 1;179(11):7827-39. doi: 10.4049/jimmunol.179.11.7827. J Immunol. 2007. PMID: 18025229
-
TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose.Pflugers Arch. 2005 Oct;451(1):212-9. doi: 10.1007/s00424-005-1446-y. Epub 2005 Jun 11. Pflugers Arch. 2005. PMID: 15952035 Review.
-
Regulation of TRPM2 channels in neutrophil granulocytes by ADP-ribose: a promising pharmacological target.Naunyn Schmiedebergs Arch Pharmacol. 2005 Apr;371(4):325-33. doi: 10.1007/s00210-005-1033-y. Naunyn Schmiedebergs Arch Pharmacol. 2005. PMID: 15841395 Review.
Cited by
-
Pore collapse underlies irreversible inactivation of TRPM2 cation channel currents.Proc Natl Acad Sci U S A. 2012 Aug 14;109(33):13440-5. doi: 10.1073/pnas.1204702109. Epub 2012 Jul 30. Proc Natl Acad Sci U S A. 2012. PMID: 22847436 Free PMC article.
-
The Molecular Basis of COVID-19 Pathogenesis, Conventional and Nanomedicine Therapy.Int J Mol Sci. 2021 May 21;22(11):5438. doi: 10.3390/ijms22115438. Int J Mol Sci. 2021. PMID: 34064039 Free PMC article. Review.
-
A TRP that makes us feel hyper.J Physiol. 2012 Apr 15;590(8):1779-80. doi: 10.1113/jphysiol.2012.229187. J Physiol. 2012. PMID: 22532645 Free PMC article. No abstract available.
-
P2X7 Receptor Triggers Lysosomal Leakage Through Calcium Mobilization in a Mechanism Dependent on Pannexin-1 Hemichannels.Front Immunol. 2022 Feb 9;13:752105. doi: 10.3389/fimmu.2022.752105. eCollection 2022. Front Immunol. 2022. PMID: 35222364 Free PMC article.
-
Mucolipins: Intracellular TRPML1-3 channels.FEBS Lett. 2010 May 17;584(10):2013-21. doi: 10.1016/j.febslet.2009.12.056. Epub 2010 Jan 13. FEBS Lett. 2010. PMID: 20074572 Free PMC article. Review.
References
-
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature. 2001;411:595–599. - PubMed
-
- Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, Furuichi K. Immunocyte Ca2+ influx system mediated by LTRPC2. Science. 2001;293:1327–1330. - PubMed
-
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–173. - PubMed
-
- McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. - PubMed
-
- Kolisek M, Beck A, Fleig A, Penner R. Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell. 2005;18:61–69. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
