

Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase
- PMID: 19443905
- PMCID: PMC2718070
- DOI: 10.1093/carcin/bgp120
Prolactin confers resistance against cisplatin in breast cancer cells by activating glutathione-S-transferase
Abstract
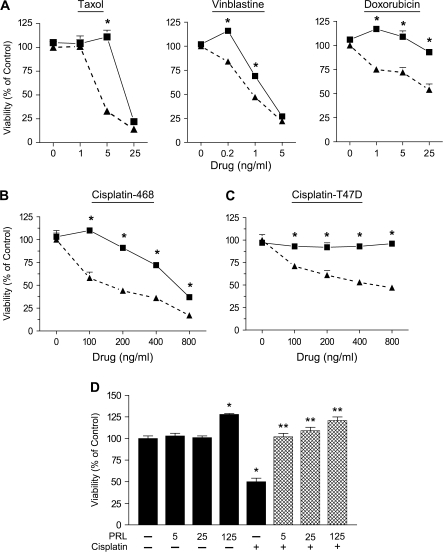
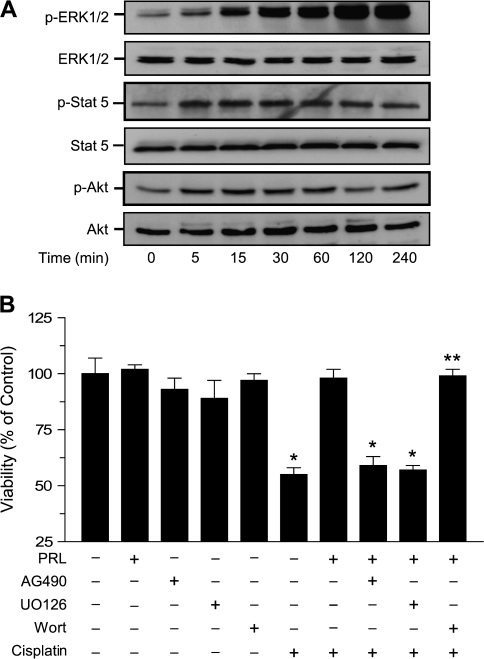
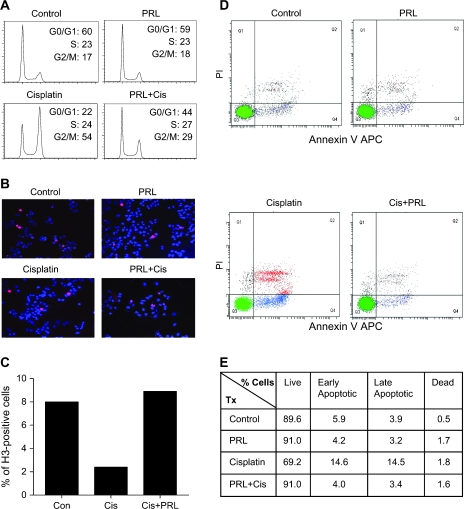
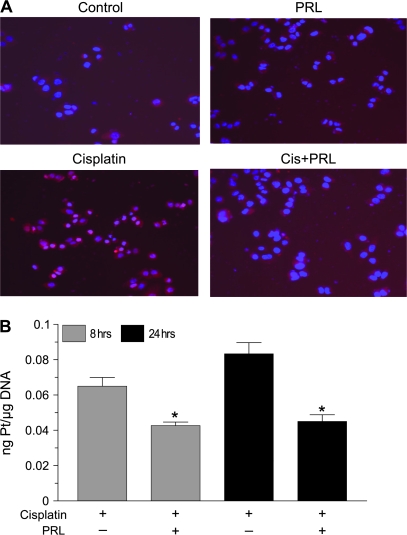
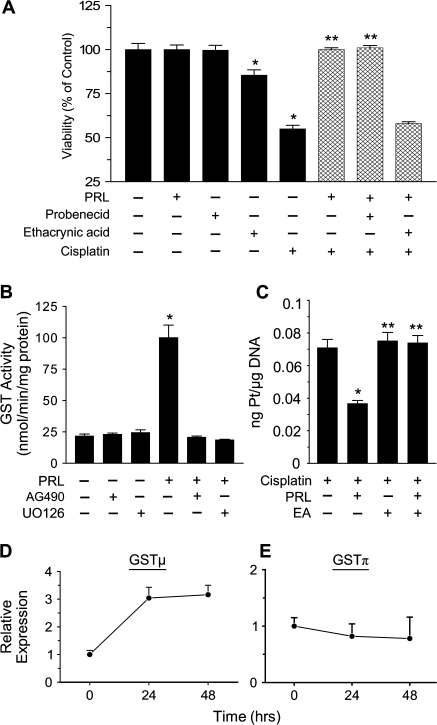
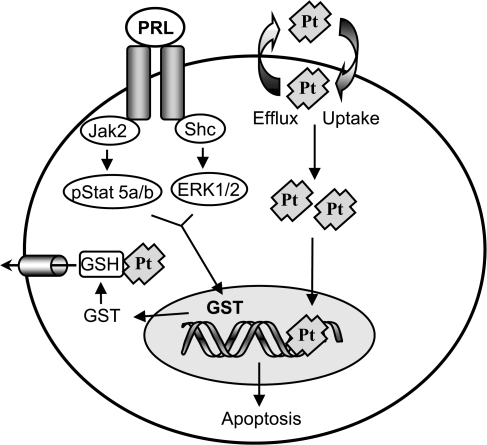
Resistance to chemotherapy is a major obstacle for successful treatment of breast cancer patients. Given that prolactin (PRL) acts as an anti-apoptotic/survival factor in the breast, we postulated that it antagonizes cytotoxicity by chemotherapeutic drugs. Treatment of breast cancer cells with PRL caused variable resistance to taxol, vinblastine, doxorubicin and cisplatin. PRL prevented cisplatin-induced G(2)/M cell cycle arrest and apoptosis. In the presence of PRL, significantly less cisplatin was bound to DNA, as determined by mass spectroscopy, and little DNA damage was seen by gamma-H2AX staining. PRL dramatically increased the activity of glutathione-S-transferase (GST), which sequesters cisplatin in the cytoplasm; this increase was abrogated by Jak and mitogen-activated protein kinase inhibitors. PRL upregulated the expression of the GSTmu, but not the pi, isozyme. A GST inhibitor abrogated antagonism of cisplatin cytotoxicity by PRL. In conclusion, PRL confers resistance against cisplatin by activating a detoxification enzyme, thereby reducing drug entry into the nucleus. These data provide a rational explanation for the ineffectiveness of cisplatin in breast cancer, which is characterized by high expression of both PRL and its receptor. Suppression of PRL production or blockade of its actions should benefit patients undergoing chemotherapy by allowing for lower drug doses and expanded drug options.
Figures
Similar articles
-
Novel roles of prolactin and estrogens in breast cancer: resistance to chemotherapy.Endocr Relat Cancer. 2010 Feb 25;17(2):R91-107. doi: 10.1677/ERC-09-0253. Print 2010 Jun. Endocr Relat Cancer. 2010. PMID: 20071456 Review.
-
Mucuna pruriens (L.) DC chemo sensitize human breast cancer cells via downregulation of prolactin-mediated JAK2/STAT5A signaling.J Ethnopharmacol. 2018 May 10;217:23-35. doi: 10.1016/j.jep.2018.02.006. Epub 2018 Feb 7. J Ethnopharmacol. 2018. PMID: 29427634
-
Pharmacological and small interference RNA-mediated inhibition of breast cancer-associated fatty acid synthase (oncogenic antigen-519) synergistically enhances Taxol (paclitaxel)-induced cytotoxicity.Int J Cancer. 2005 May 20;115(1):19-35. doi: 10.1002/ijc.20754. Int J Cancer. 2005. PMID: 15657900
-
MicroRNA-133b targets glutathione S-transferase π expression to increase ovarian cancer cell sensitivity to chemotherapy drugs.Drug Des Devel Ther. 2015 Sep 16;9:5225-35. doi: 10.2147/DDDT.S87526. eCollection 2015. Drug Des Devel Ther. 2015. PMID: 26396496 Free PMC article.
-
Effectivity of Bromocriptine Administration Towards Prolactin Positive Breast Cancer Receiving Anthracycline-Based Chemotherapy: A Literature Review.Acta Med Indones. 2023 Oct;55(4):465-474. Acta Med Indones. 2023. PMID: 38213041 Review.
Cited by
-
Expression of glutathione, glutathione peroxidase and glutathione S-transferase pi in canine mammary tumors.BMC Vet Res. 2014 Feb 24;10:49. doi: 10.1186/1746-6148-10-49. BMC Vet Res. 2014. PMID: 24565113 Free PMC article. Clinical Trial.
-
Prolactin: The Third Hormone in Breast Cancer.Front Endocrinol (Lausanne). 2022 Jun 16;13:910978. doi: 10.3389/fendo.2022.910978. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 35784527 Free PMC article. Review.
-
Lactogens and estrogens in breast cancer chemoresistance.Expert Rev Endocrinol Metab. 2011 May;6(3):411-422. doi: 10.1586/eem.11.19. Expert Rev Endocrinol Metab. 2011. PMID: 21731573 Free PMC article.
-
Prolactin and its receptor as therapeutic targets in glioblastoma multiforme.Sci Rep. 2019 Dec 20;9(1):19578. doi: 10.1038/s41598-019-55860-x. Sci Rep. 2019. PMID: 31862900 Free PMC article.
-
Identifying novel hypoxia-associated markers of chemoresistance in ovarian cancer.BMC Cancer. 2015 Jul 25;15:547. doi: 10.1186/s12885-015-1539-8. BMC Cancer. 2015. PMID: 26205780 Free PMC article.
References
-
- Parkin DM, et al. Use of statistics to assess the global burden of breast cancer. Breast J. 2006;12(suppl. 1):S70–S80. - PubMed
-
- Decatris MP, et al. Platinum-based chemotherapy in metastatic breast cancer: current status. Cancer Treat. Rev. 2004;30:53–81. - PubMed
-
- Wang D, et al. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug Discov. 2005;4:307–320. - PubMed
-
- Coley HM. Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat. Rev. 2008;34:378–390. - PubMed
-
- Igney FH, et al. Death and anti-death: tumour resistance to apoptosis. Nat. Rev. Cancer. 2002;2:277–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
