

A partial form of recessive STAT1 deficiency in humans
- PMID: 19436109
- PMCID: PMC2689115
- DOI: 10.1172/JCI37083
A partial form of recessive STAT1 deficiency in humans
Abstract
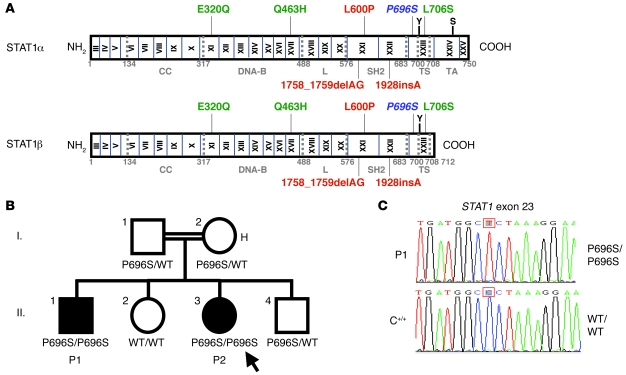
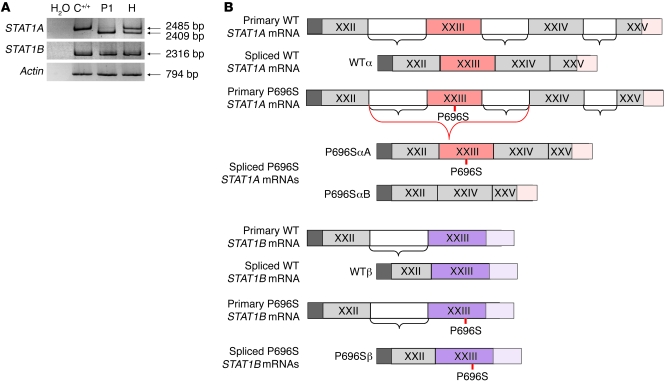
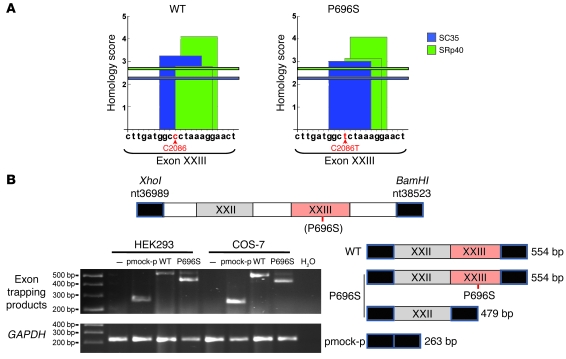
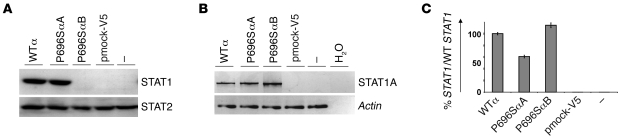
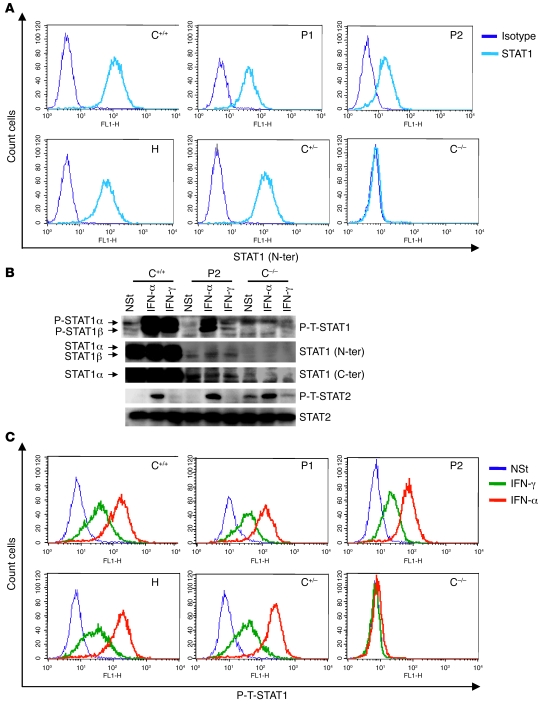
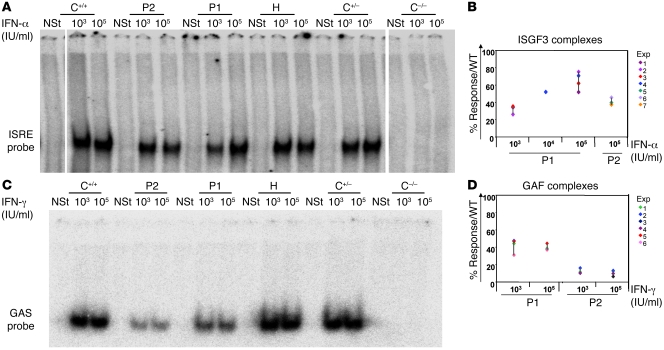
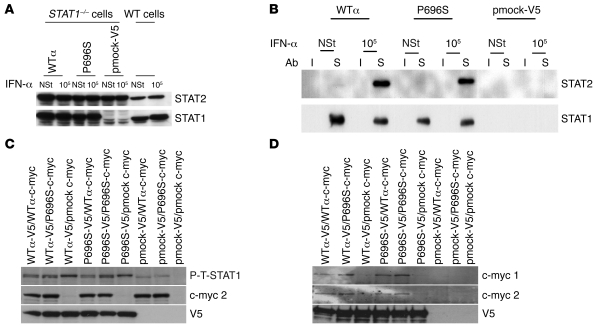
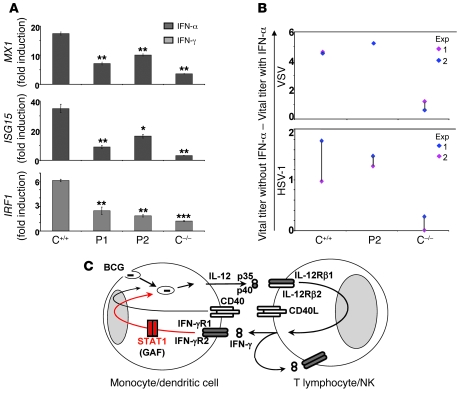
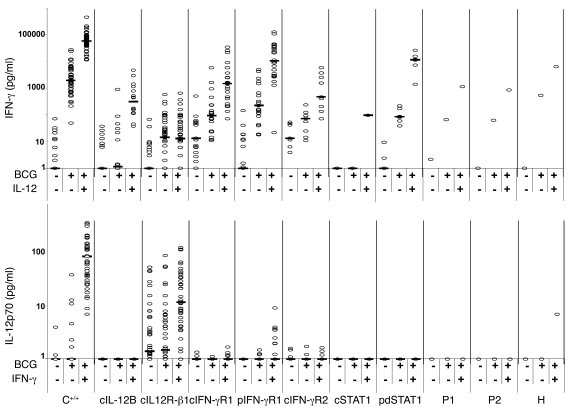
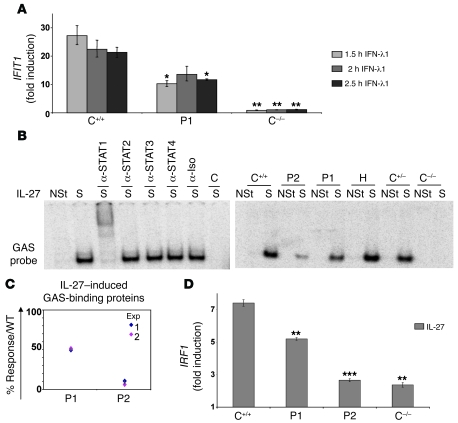
Complete STAT1 deficiency is an autosomal recessive primary immunodeficiency caused by null mutations that abolish STAT1-dependent cellular responses to both IFN-alpha/beta and IFN-gamma. Affected children suffer from lethal intracellular bacterial and viral diseases. Here we report a recessive form of partial STAT1 deficiency, characterized by impaired but not abolished IFN-alpha/beta and IFN-gamma signaling. Two affected siblings suffered from severe but curable intracellular bacterial and viral diseases. Both were homozygous for a missense STAT1 mutation: g.C2086T (P696S). This STAT1 allele impaired the splicing of STAT1 mRNA, probably by disrupting an exonic splice enhancer. The misspliced forms were not translated into a mature protein. The allele was hypofunctional, because residual full-length mRNA production resulted in low but detectable levels of normally functional STAT1 proteins. The P696S amino acid substitution was not detrimental. The patients' cells, therefore, displayed impaired but not abolished responses to both IFN-alpha and IFN-gamma. We also show that recessive STAT1 deficiencies impaired the IL-27 and IFN-lambda1 signaling pathways, possibly contributing to the predisposition to bacterial and viral infections, respectively. Partial recessive STAT1 deficiency is what we believe to be a novel primary immunodeficiency, resulting in impairment of the response to at least 4 cytokines (IFN-alpha/beta, IFN-gamma, IFN-lambda1, and IL-27). It should be considered in patients with unexplained, severe, but curable intracellular bacterial and viral infections.
Figures
Similar articles
-
A novel form of human STAT1 deficiency impairing early but not late responses to interferons.Blood. 2010 Dec 23;116(26):5895-906. doi: 10.1182/blood-2010-04-280586. Epub 2010 Sep 14. Blood. 2010. PMID: 20841510 Free PMC article.
-
Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis.J Exp Med. 2011 Aug 1;208(8):1635-48. doi: 10.1084/jem.20110958. Epub 2011 Jul 4. J Exp Med. 2011. PMID: 21727188 Free PMC article.
-
Severe impairment of IFN-γ and IFN-α responses in cells of a patient with a novel STAT1 splicing mutation.Blood. 2011 Aug 18;118(7):1806-17. doi: 10.1182/blood-2011-01-330571. Epub 2011 Jul 19. Blood. 2011. PMID: 21772053
-
Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant.Pediatr Allergy Immunol. 2021 Nov;32(8):1804-1812. doi: 10.1111/pai.13603. Epub 2021 Aug 9. Pediatr Allergy Immunol. 2021. PMID: 34289170 Review.
-
Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes.Curr Opin Immunol. 2012 Aug;24(4):364-78. doi: 10.1016/j.coi.2012.04.011. Epub 2012 May 30. Curr Opin Immunol. 2012. PMID: 22651901 Free PMC article. Review.
Cited by
-
Inborn errors of immunity with loss- and gain-of-function germline mutations in STAT1.Clin Exp Immunol. 2023 Apr 25;212(2):96-106. doi: 10.1093/cei/uxac106. Clin Exp Immunol. 2023. PMID: 36420581 Free PMC article. Review.
-
Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity.Semin Immunol. 2014 Dec;26(6):454-70. doi: 10.1016/j.smim.2014.09.008. Epub 2014 Oct 26. Semin Immunol. 2014. PMID: 25453225 Free PMC article. Review.
-
High throughput screening for small molecule enhancers of the interferon signaling pathway to drive next-generation antiviral drug discovery.PLoS One. 2012;7(5):e36594. doi: 10.1371/journal.pone.0036594. Epub 2012 May 4. PLoS One. 2012. PMID: 22574190 Free PMC article.
-
Live Cell Imaging Demonstrates Multiple Routes Toward a STAT1 Gain-of-Function Phenotype.Front Immunol. 2020 Jun 9;11:1114. doi: 10.3389/fimmu.2020.01114. eCollection 2020. Front Immunol. 2020. PMID: 32582194 Free PMC article.
-
Immunity to infection in IL-17-deficient mice and humans.Eur J Immunol. 2012 Sep;42(9):2246-54. doi: 10.1002/eji.201242605. Eur J Immunol. 2012. PMID: 22949323 Free PMC article. Review.
References
-
- Alvarez G.R., Zwilling B.S., Lafuse W.P. Mycobacterium avium inhibition of IFN-gamma signaling in mouse macrophages: Toll-like receptor 2 stimulation increases expression of dominant-negative STAT1 beta by mRNA stabilization. J. Immunol. 2003;171:6766–6773. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
