

Avoiding the high Bonferroni penalty in genome-wide association studies
- PMID: 19434714
- PMCID: PMC2796708
- DOI: 10.1002/gepi.20430
Avoiding the high Bonferroni penalty in genome-wide association studies
Abstract
A major challenge in genome-wide association studies (GWASs) is to derive the multiple testing threshold when hypothesis tests are conducted using a large number of single nucleotide polymorphisms. Permutation tests are considered the gold standard in multiple testing adjustment in genetic association studies. However, it is computationally intensive, especially for GWASs, and can be impractical if a large number of random shuffles are used to ensure accuracy. Many researchers have developed approximation algorithms to relieve the computing burden imposed by permutation. One particularly attractive alternative to permutation is to calculate the effective number of independent tests, M(eff), which has been shown to be promising in genetic association studies. In this study, we compare recently developed M(eff) methods and validate them by the permutation test with 10,000 random shuffles using two real GWAS data sets: an Illumina 1M BeadChip and an Affymetrix GeneChip Human Mapping 500K Array Set. Our results show that the simpleM method produces the best approximation of the permutation threshold, and it does so in the shortest amount of time. We also show that M(eff) is indeed valid on a genome-wide scale in these data sets based on statistical theory and significance tests. The significance thresholds derived can provide practical guidelines for other studies using similar population samples and genotyping platforms.
Figures
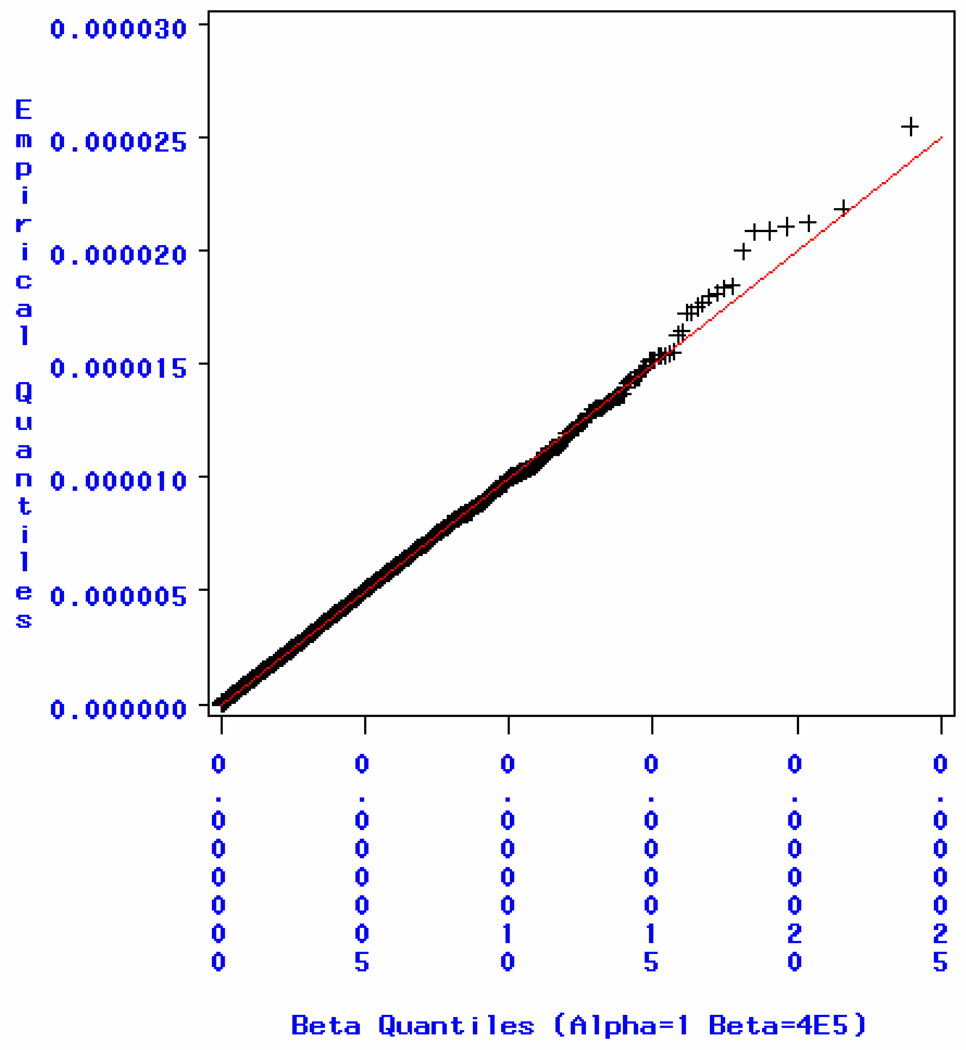
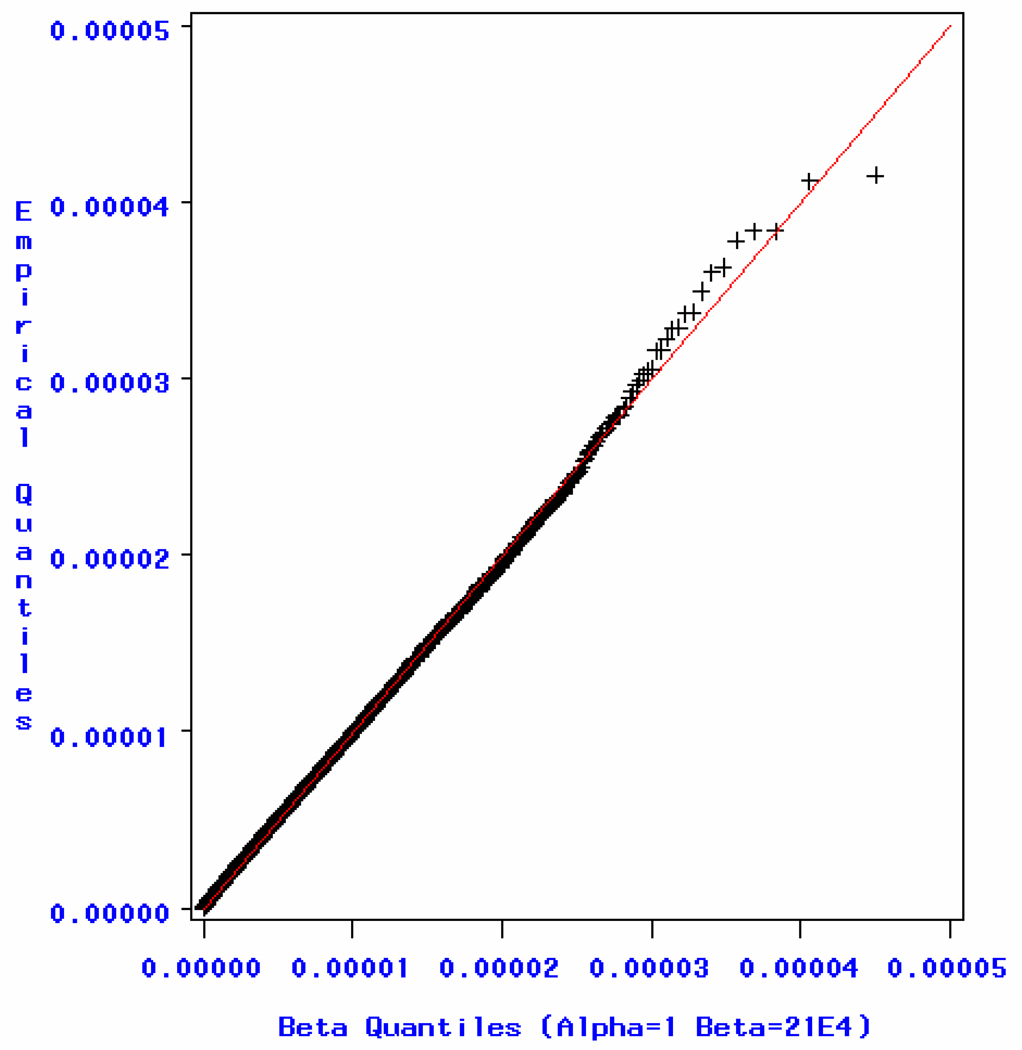
Similar articles
-
Multiple testing corrections for imputed SNPs.Genet Epidemiol. 2011 Apr;35(3):154-8. doi: 10.1002/gepi.20563. Epub 2011 Jan 19. Genet Epidemiol. 2011. PMID: 21254223 Free PMC article.
-
Factors affecting the effective number of tests in genetic association studies: a comparative study of three PCA-based methods.J Hum Genet. 2011 Jun;56(6):428-35. doi: 10.1038/jhg.2011.34. Epub 2011 Mar 31. J Hum Genet. 2011. PMID: 21451529
-
Batch effects in the BRLMM genotype calling algorithm influence GWAS results for the Affymetrix 500K array.Pharmacogenomics J. 2010 Aug;10(4):336-46. doi: 10.1038/tpj.2010.36. Pharmacogenomics J. 2010. PMID: 20676071
-
Missing data imputation and haplotype phase inference for genome-wide association studies.Hum Genet. 2008 Dec;124(5):439-50. doi: 10.1007/s00439-008-0568-7. Epub 2008 Oct 11. Hum Genet. 2008. PMID: 18850115 Free PMC article. Review.
-
The extent of linkage disequilibrium and computational challenges of single nucleotide polymorphisms in genome-wide association studies.Curr Drug Metab. 2011 Jun;12(5):498-506. doi: 10.2174/138920011795495312. Curr Drug Metab. 2011. PMID: 21453276 Review.
Cited by
-
Multiple testing correction in linear mixed models.Genome Biol. 2016 Apr 1;17:62. doi: 10.1186/s13059-016-0903-6. Genome Biol. 2016. PMID: 27039378 Free PMC article.
-
Genetic loci associated with circulating phospholipid trans fatty acids: a meta-analysis of genome-wide association studies from the CHARGE Consortium.Am J Clin Nutr. 2015 Feb;101(2):398-406. doi: 10.3945/ajcn.114.094557. Epub 2014 Dec 10. Am J Clin Nutr. 2015. PMID: 25646338 Free PMC article.
-
A genome-wide scan for breast cancer risk haplotypes among African American women.PLoS One. 2013;8(2):e57298. doi: 10.1371/journal.pone.0057298. Epub 2013 Feb 28. PLoS One. 2013. PMID: 23468962 Free PMC article.
-
Heterozygosity Ratio, a Robust Global Genomic Measure of Autozygosity and Its Association with Height and Disease Risk.Genetics. 2016 Nov;204(3):893-904. doi: 10.1534/genetics.116.189936. Epub 2016 Aug 31. Genetics. 2016. PMID: 27585849 Free PMC article.
-
Common Genetic Variation in CYP17A1 and Response to Abiraterone Acetate in Patients with Metastatic Castration-Resistant Prostate Cancer.Int J Mol Sci. 2016 Jul 9;17(7):1097. doi: 10.3390/ijms17071097. Int J Mol Sci. 2016. PMID: 27409606 Free PMC article.
References
-
- Armitage P. Tests for Linear Trends in Proportions and Frequencies. Biometrics. 1955;11(3):375–386.
-
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. - PubMed
-
- Cheverud JM. A simple correction for multiple comparisons in interval mapping genome scans. Heredity. 2001;87(1):52–58. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 HL087698-03/HL/NHLBI NIH HHS/United States
- HL088655/HL/NHLBI NIH HHS/United States
- HL087700/HL/NHLBI NIH HHS/United States
- T32 ES007126/ES/NIEHS NIH HHS/United States
- 1R01HL08768801/HL/NHLBI NIH HHS/United States
- R01 HL088215/HL/NHLBI NIH HHS/United States
- HL088215/HL/NHLBI NIH HHS/United States
- U01 AG023746/AG/NIA NIH HHS/United States
- R01 HL087700/HL/NHLBI NIH HHS/United States
- AG023746/AG/NIA NIH HHS/United States
- 5R01HL08770002/HL/NHLBI NIH HHS/United States
- 5R01HL087698/HL/NHLBI NIH HHS/United States
- U01 HL088655/HL/NHLBI NIH HHS/United States
- R01 HL087698/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
