

Bmi1 regulates mitochondrial function and the DNA damage response pathway
- PMID: 19404261
- PMCID: PMC4721521
- DOI: 10.1038/nature08040
Bmi1 regulates mitochondrial function and the DNA damage response pathway
Abstract
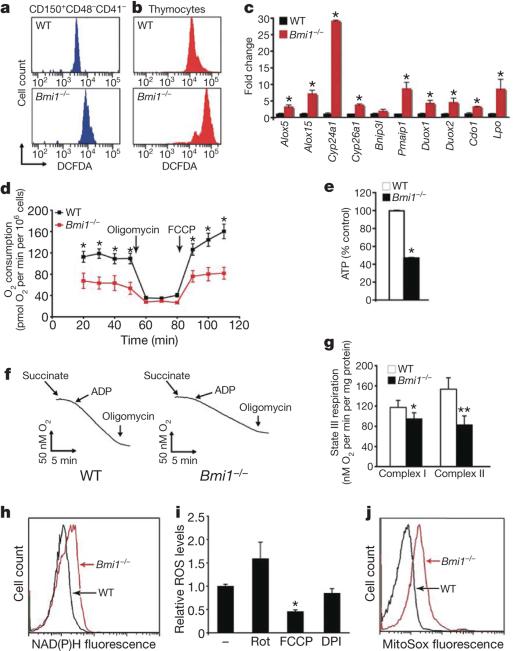
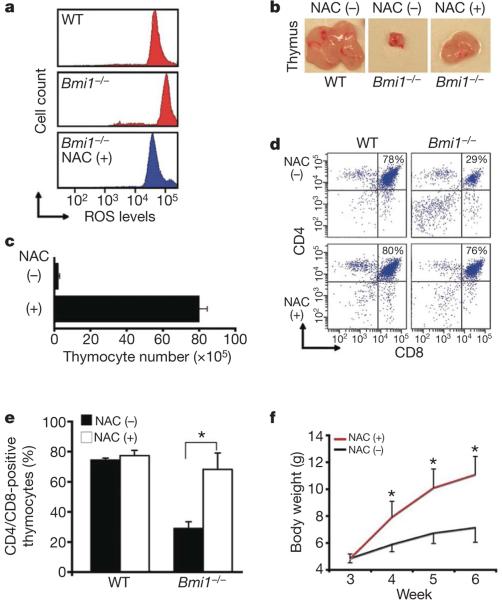
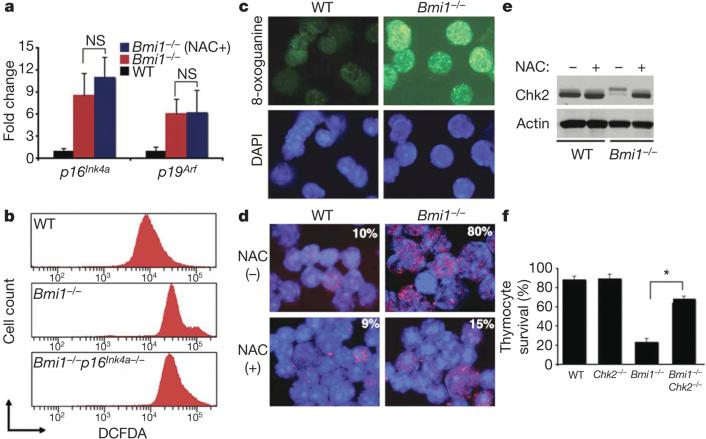
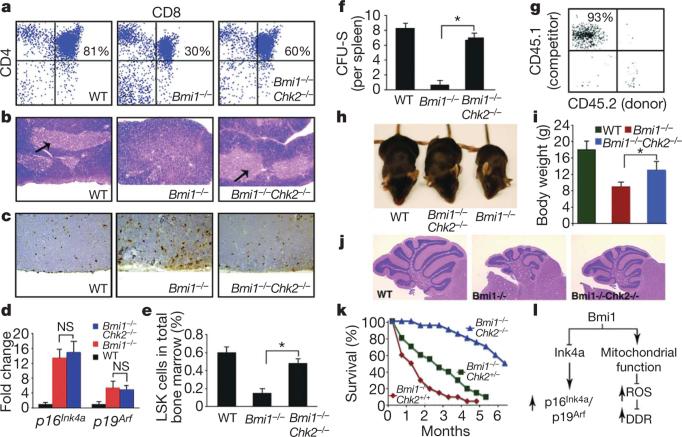
Mice deficient in the Polycomb repressor Bmi1 develop numerous abnormalities including a severe defect in stem cell self-renewal, alterations in thymocyte maturation and a shortened lifespan. Previous work has implicated de-repression of the Ink4a/Arf (also known as Cdkn2a) locus as mediating many of the aspects of the Bmi1(-/-) phenotype. Here we demonstrate that cells derived from Bmi1(-/-) mice also have impaired mitochondrial function, a marked increase in the intracellular levels of reactive oxygen species and subsequent engagement of the DNA damage response pathway. Furthermore, many of the deficiencies normally observed in Bmi1(-/-) mice improve after either pharmacological treatment with the antioxidant N-acetylcysteine or genetic disruption of the DNA damage response pathway by Chk2 (also known as Chek2) deletion. These results demonstrate that Bmi1 has an unexpected role in maintaining mitochondrial function and redox homeostasis and indicate that the Polycomb family of proteins can coordinately regulate cellular metabolism with stem and progenitor cell function.
Figures
Similar articles
-
BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery.J Neurosci. 2010 Jul 28;30(30):10096-111. doi: 10.1523/JNEUROSCI.1634-10.2010. J Neurosci. 2010. PMID: 20668194 Free PMC article.
-
Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma.Cancer Cell. 2007 Oct;12(4):328-41. doi: 10.1016/j.ccr.2007.08.032. Cancer Cell. 2007. PMID: 17936558
-
Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both Ink4a/Arf-dependent and -independent manners in mice.Hepatology. 2010 Sep;52(3):1111-23. doi: 10.1002/hep.23793. Hepatology. 2010. PMID: 20648475
-
Role of the polycomb group gene BMI1 in normal and leukemic hematopoietic stem and progenitor cells.Curr Opin Hematol. 2010 Jul;17(4):294-9. doi: 10.1097/MOH.0b013e328338c439. Curr Opin Hematol. 2010. PMID: 20308890 Review.
-
BMI1 as a novel target for drug discovery in cancer.J Cell Biochem. 2011 Oct;112(10):2729-41. doi: 10.1002/jcb.23234. J Cell Biochem. 2011. PMID: 21678481 Review.
Cited by
-
Nuclear-encoded cytochrome c oxidase subunit 4 regulates BMI1 expression and determines proliferative capacity of high-grade gliomas.Oncotarget. 2015 Feb 28;6(6):4330-44. doi: 10.18632/oncotarget.3015. Oncotarget. 2015. PMID: 25726526 Free PMC article.
-
Stress and stem cells.Wiley Interdiscip Rev Dev Biol. 2012 Nov-Dec;1(6):789-802. doi: 10.1002/wdev.56. Epub 2012 Apr 18. Wiley Interdiscip Rev Dev Biol. 2012. PMID: 23799624 Free PMC article.
-
CD44, Hyaluronan, the Hematopoietic Stem Cell, and Leukemia-Initiating Cells.Front Immunol. 2015 May 26;6:235. doi: 10.3389/fimmu.2015.00235. eCollection 2015. Front Immunol. 2015. PMID: 26074915 Free PMC article. Review.
-
Reduced TRMU expression increases the sensitivity of hair-cell-like HEI-OC-1 cells to neomycin damage in vitro.Sci Rep. 2016 Jul 13;6:29621. doi: 10.1038/srep29621. Sci Rep. 2016. PMID: 27405449 Free PMC article.
-
BMI1 confers radioresistance to normal and cancerous neural stem cells through recruitment of the DNA damage response machinery.J Neurosci. 2010 Jul 28;30(30):10096-111. doi: 10.1523/JNEUROSCI.1634-10.2010. J Neurosci. 2010. PMID: 20668194 Free PMC article.
References
-
- Park IK, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. - PubMed
-
- van der Lugt NM, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 protooncogene. Genes Dev. 1994;8:757–769. - PubMed
-
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
