

Trypsin-ligand binding free energies from explicit and implicit solvent simulations with polarizable potential
- PMID: 19399779
- PMCID: PMC2752704
- DOI: 10.1002/jcc.21268
Trypsin-ligand binding free energies from explicit and implicit solvent simulations with polarizable potential
Abstract
We have calculated the binding free energies of a series of benzamidine-like inhibitors to trypsin with a polarizable force field using both explicit and implicit solvent approaches. Free energy perturbation has been performed for the ligands in bulk water and in protein complex with molecular dynamics simulations. The binding free energies calculated from explicit solvent simulations are well within the accuracy of experimental measurement and the direction of change is predicted correctly in all cases. We analyzed the molecular dipole moments of the ligands in gas, water and protein environments. Neither binding affinity nor ligand solvation free energy in bulk water shows much dependence on the molecular dipole moments of the ligands. Substitution of the aromatic or the charged group in the ligand results in considerable change in the solvation energy in bulk water and protein whereas the binding affinity varies insignificantly due to cancellation. The effect of chemical modification on ligand charge distribution is mostly local. Replacing benzene with diazine has minimal impact on the atomic multipoles at the amidinium group. We have also utilized an implicit solvent based end-state approach to evaluate the binding free energies of these inhibitors. In this approach, the polarizable multipole model combined with Poisson-Boltzmann/surface area (PMPB/SA) provides the electrostatic interaction energy and the polar solvation free energy. Overall the relative binding free energies obtained from the MM-PMPB/SA model are in good agreement with the experimental data.
2009 Wiley Periodicals, Inc.
Figures
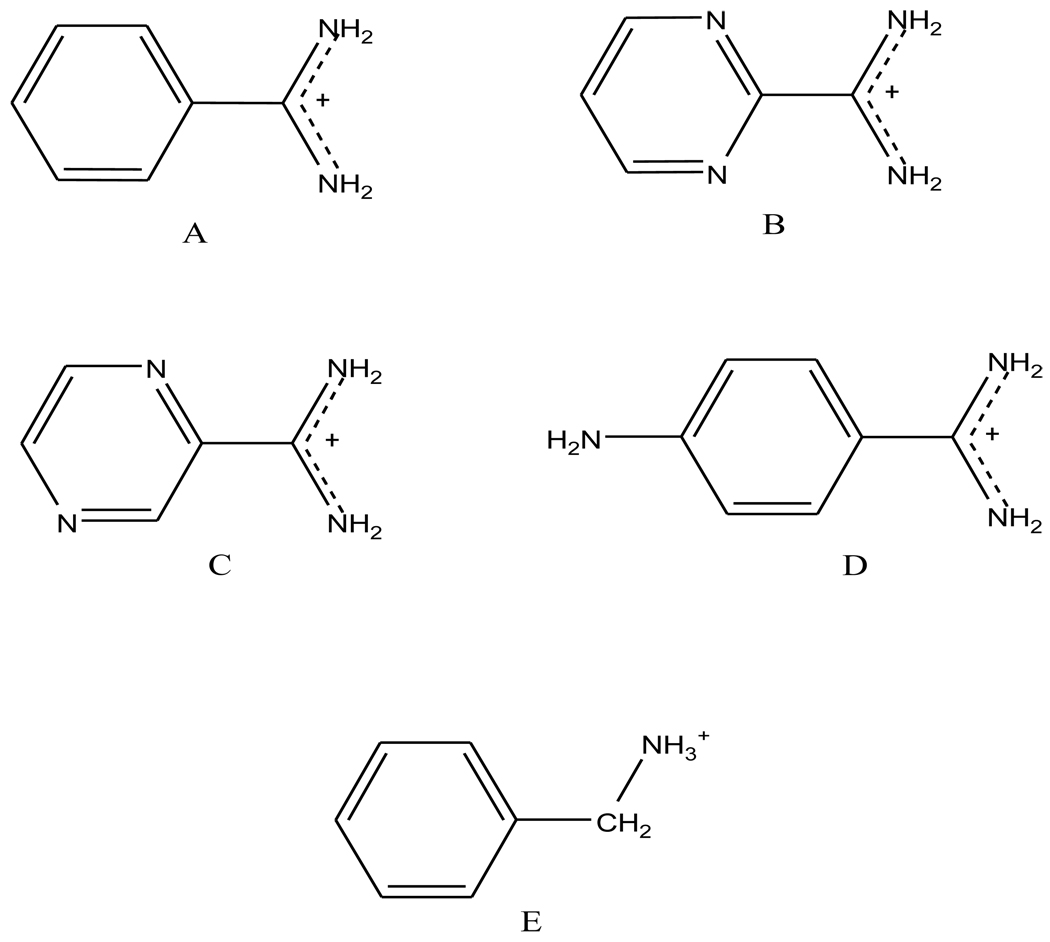
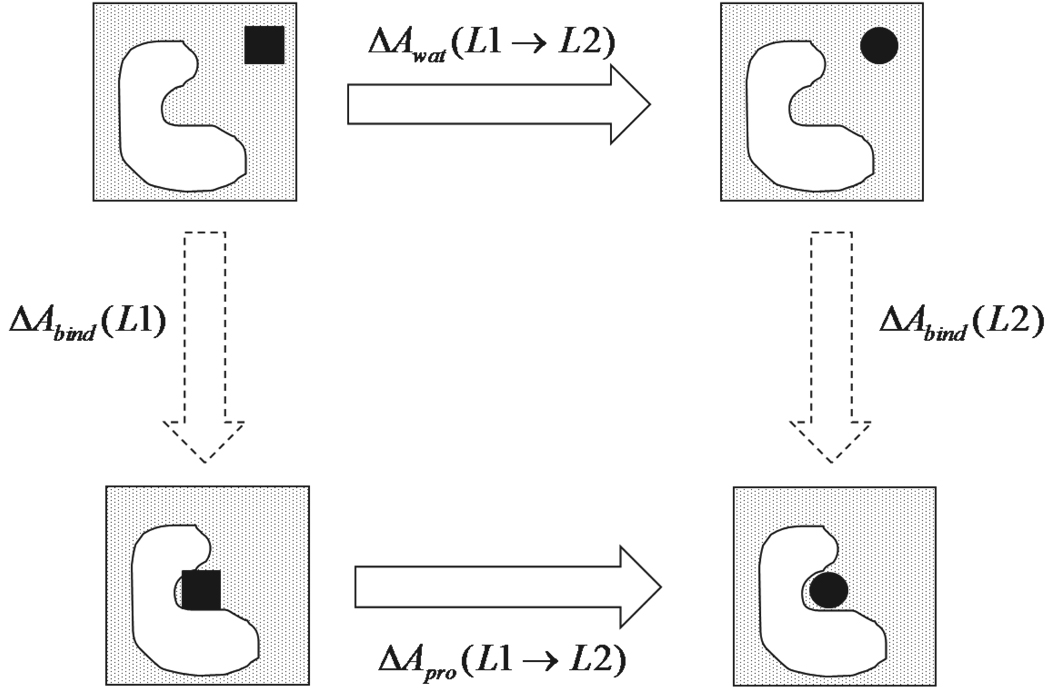
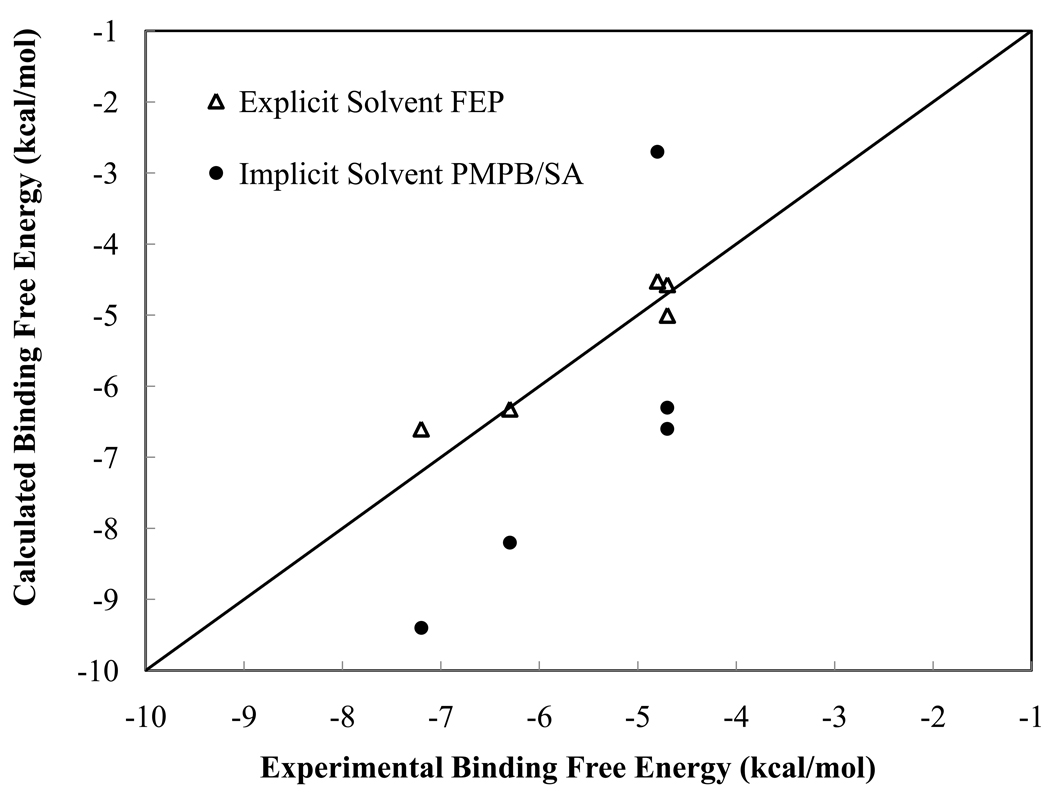
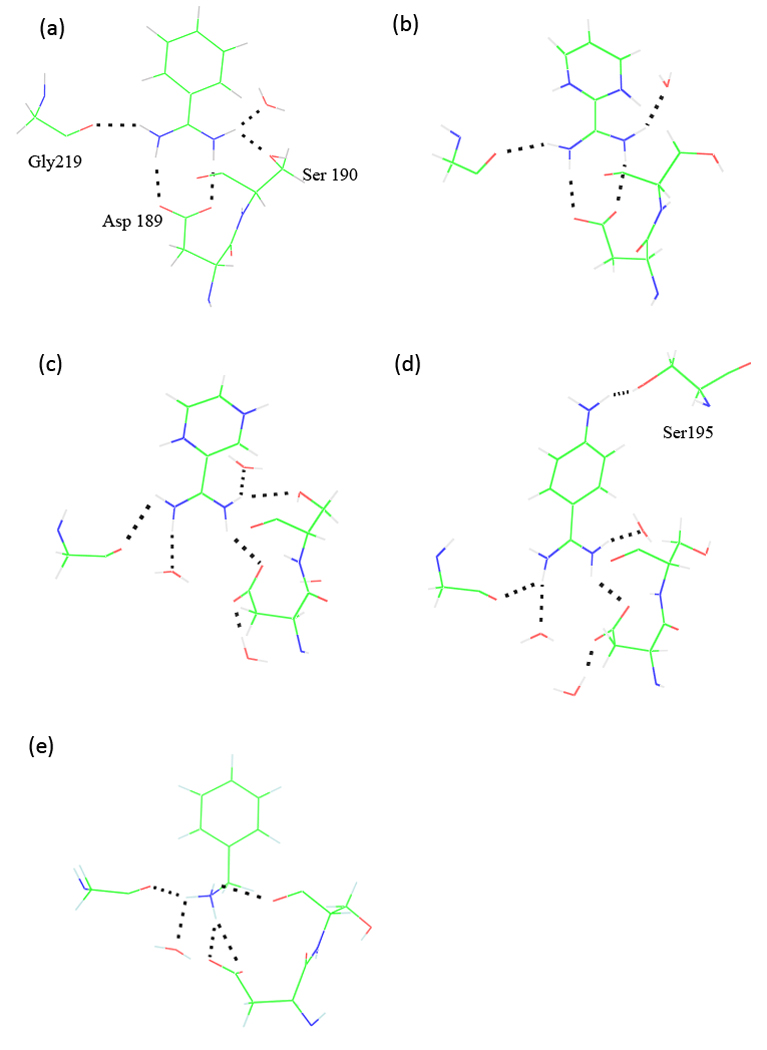
Similar articles
-
Trypsin-ligand binding free energy calculation with AMOEBA.Annu Int Conf IEEE Eng Med Biol Soc. 2009;2009:2328-31. doi: 10.1109/IEMBS.2009.5335108. Annu Int Conf IEEE Eng Med Biol Soc. 2009. PMID: 19965178 Free PMC article.
-
Calculation of protein-ligand binding free energy by using a polarizable potential.Proc Natl Acad Sci U S A. 2008 Apr 29;105(17):6290-5. doi: 10.1073/pnas.0711686105. Epub 2008 Apr 21. Proc Natl Acad Sci U S A. 2008. PMID: 18427113 Free PMC article.
-
Receptor rigidity and ligand mobility in trypsin-ligand complexes.Proteins. 2005 Feb 1;58(2):407-17. doi: 10.1002/prot.20326. Proteins. 2005. PMID: 15578663
-
Polarizable atomic multipole solutes in a Poisson-Boltzmann continuum.J Chem Phys. 2007 Mar 28;126(12):124114. doi: 10.1063/1.2714528. J Chem Phys. 2007. PMID: 17411115 Free PMC article. Review.
-
Computer simulations with explicit solvent: recent progress in the thermodynamic decomposition of free energies and in modeling electrostatic effects.Annu Rev Phys Chem. 1998;49:531-67. doi: 10.1146/annurev.physchem.49.1.531. Annu Rev Phys Chem. 1998. PMID: 9933909 Review.
Cited by
-
Probing the effect of conformational constraint on phosphorylated ligand binding to an SH2 domain using polarizable force field simulations.J Phys Chem B. 2012 Feb 9;116(5):1716-27. doi: 10.1021/jp210265d. Epub 2012 Jan 31. J Phys Chem B. 2012. PMID: 22214214 Free PMC article.
-
Trypsin-ligand binding free energy calculation with AMOEBA.Annu Int Conf IEEE Eng Med Biol Soc. 2009;2009:2328-31. doi: 10.1109/IEMBS.2009.5335108. Annu Int Conf IEEE Eng Med Biol Soc. 2009. PMID: 19965178 Free PMC article.
-
Virtual screening using molecular simulations.Proteins. 2011 Jun;79(6):1940-51. doi: 10.1002/prot.23018. Epub 2011 Apr 12. Proteins. 2011. PMID: 21491494 Free PMC article.
-
Drude polarizable force field for aliphatic ketones and aldehydes, and their associated acyclic carbohydrates.J Comput Aided Mol Des. 2017 Apr;31(4):349-363. doi: 10.1007/s10822-017-0010-0. Epub 2017 Feb 11. J Comput Aided Mol Des. 2017. PMID: 28190218 Free PMC article.
-
Computational investigation of O2 diffusion through an intra-molecular tunnel in AlkB; influence of polarization on O2 transport.Chem Sci. 2017 Sep 1;8(9):6230-6238. doi: 10.1039/c7sc00997f. Epub 2017 Jul 5. Chem Sci. 2017. PMID: 28989656 Free PMC article.
References
-
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Accounts Chem Res. 2000;33(12):889–897. - PubMed
-
- Gohlke H, Klebe G. Angew Chem Int Edit. 2002;41(15):2645–2676. - PubMed
-
- Brandsdal BO, Osterberg F, Almlof M, Feierberg I, Luzhkov VB, Aqvist J. Adv Protein Chem. 2003;66:123. - PubMed
-
- Jorgensen WL. Science. 2004;303(5665):1813–1818. - PubMed
-
- Gilson MK, Zhou HX. Annual Review of Biophysics and Biomolecular Structure. 2007;36:21–42. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
