

GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA
- PMID: 19339690
- PMCID: PMC2666342
- DOI: 10.1101/gad.1748409
GCN5 is a required cofactor for a ubiquitin ligase that targets NF-kappaB/RelA
Abstract
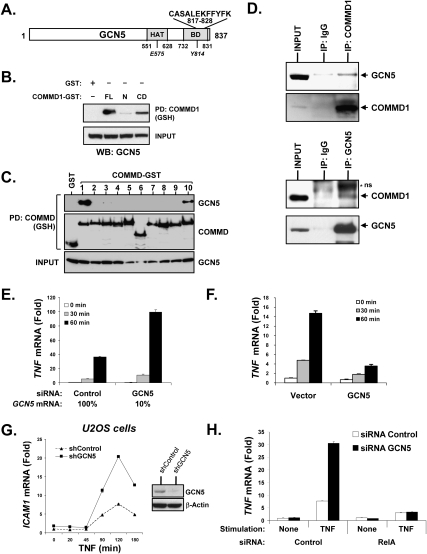
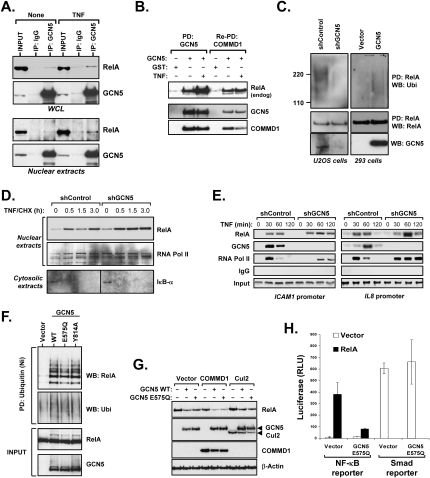
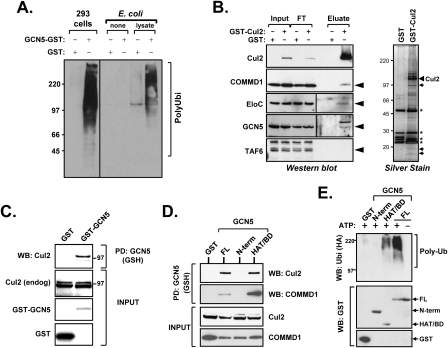
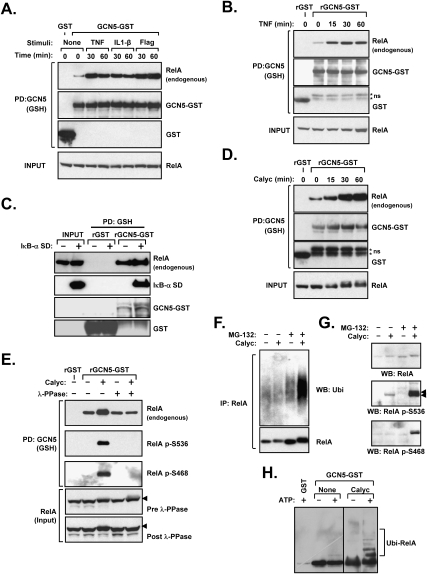
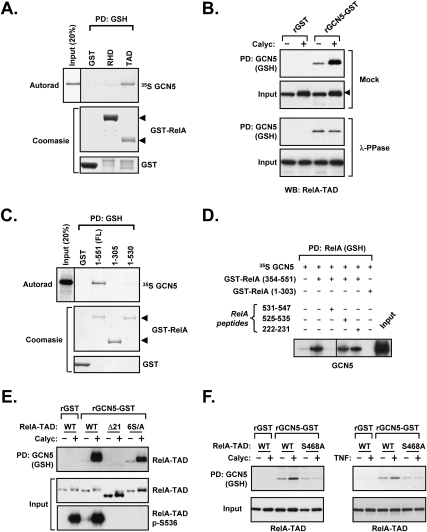
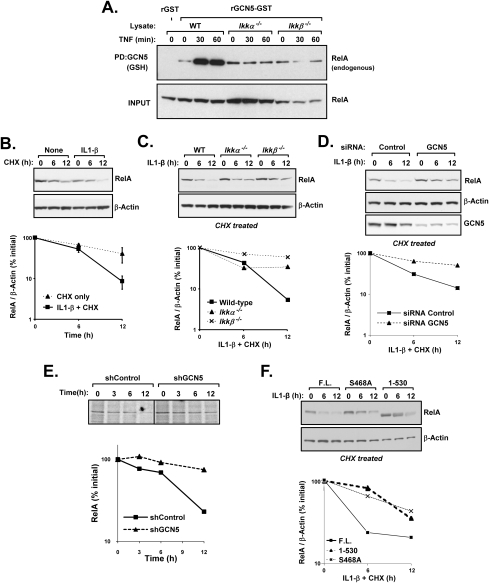
The transcription factor NF-kappaB is a critical regulator of inflammatory and cell survival signals. Proteasomal degradation of NF-kappaB subunits plays an important role in the termination of NF-kappaB activity, and at least one of the identified ubiquitin ligases is a multimeric complex containing Copper Metabolism Murr1 Domain 1 (COMMD1) and Cul2. We report here that GCN5, a histone acetyltransferase, associates with COMMD1 and other components of the ligase, promotes RelA ubiquitination, and represses kappaB-dependent transcription. In this role, the acetyltransferase activity of GCN5 is not required. Interestingly, GCN5 binds more avidly to RelA after phosphorylation on Ser 468, an event that is dependent on IKK activity. Consistent with this, we find that both GCN5 and the IkappaB Kinase (IKK) complex promote RelA degradation. Collectively, the data indicate that GCN5 participates in the ubiquitination process as an accessory factor for a ubiquitin ligase, where it provides a novel link between phosphorylation and ubiquitination.
Figures
Similar articles
-
COMMD1 promotes the ubiquitination of NF-kappaB subunits through a cullin-containing ubiquitin ligase.EMBO J. 2007 Jan 24;26(2):436-47. doi: 10.1038/sj.emboj.7601489. Epub 2006 Dec 21. EMBO J. 2007. PMID: 17183367 Free PMC article.
-
The ubiquitin ligase HERC3 attenuates NF-κB-dependent transcription independently of its enzymatic activity by delivering the RelA subunit for degradation.Nucleic Acids Res. 2015 Nov 16;43(20):9889-904. doi: 10.1093/nar/gkv1064. Epub 2015 Oct 17. Nucleic Acids Res. 2015. PMID: 26476452 Free PMC article.
-
ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment.Nucleic Acids Res. 2014 Jul;42(13):8416-32. doi: 10.1093/nar/gku529. Epub 2014 Jun 23. Nucleic Acids Res. 2014. PMID: 24957606 Free PMC article.
-
Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity.Annu Rev Immunol. 2000;18:621-63. doi: 10.1146/annurev.immunol.18.1.621. Annu Rev Immunol. 2000. PMID: 10837071 Review.
-
Nuclear ubiquitin ligases, NF-kappaB degradation, and the control of inflammation.Sci Signal. 2008 Jan 8;1(1):pe1. doi: 10.1126/stke.11pe1. Sci Signal. 2008. PMID: 18270169 Review.
Cited by
-
Deubiquitination of NF-κB by Ubiquitin-Specific Protease-7 promotes transcription.Proc Natl Acad Sci U S A. 2013 Jan 8;110(2):618-23. doi: 10.1073/pnas.1208446110. Epub 2012 Dec 24. Proc Natl Acad Sci U S A. 2013. PMID: 23267096 Free PMC article.
-
FunDMDeep-m6A: identification and prioritization of functional differential m6A methylation genes.Bioinformatics. 2019 Jul 15;35(14):i90-i98. doi: 10.1093/bioinformatics/btz316. Bioinformatics. 2019. PMID: 31510685 Free PMC article.
-
Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway.Cell Signal. 2010 Sep;22(9):1282-90. doi: 10.1016/j.cellsig.2010.03.017. Epub 2010 Apr 2. Cell Signal. 2010. PMID: 20363318 Free PMC article. Review.
-
The Direct and Indirect Roles of NF-κB in Cancer: Lessons from Oncogenic Fusion Proteins and Knock-in Mice.Biomedicines. 2018 Mar 19;6(1):36. doi: 10.3390/biomedicines6010036. Biomedicines. 2018. PMID: 29562713 Free PMC article. Review.
-
Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription.PLoS Biol. 2013 Sep;11(9):e1001647. doi: 10.1371/journal.pbio.1001647. Epub 2013 Sep 3. PLoS Biol. 2013. PMID: 24019758 Free PMC article.
References
-
- Arenzana-Seisdedos F., Turpin P., Rodriguez M., Thomas D., Hay R.T., Virelizier J.L., Dargemont C. Nuclear localization of IκBα promotes active transport of NF−κB from the nucleus to the cytoplasm. J. Cell Sci. 1997;110:369–378. - PubMed
-
- Baeuerle P.A., Baltimore D. I κ B: A specific inhibitor of the NF-κ B transcription factor. Science. 1988;242:540–546. - PubMed
-
- Burstein E., Hoberg J.E., Wilkinson A.S., Rumble J.M., Csomos R.A., Komarck C.M., Maine G.N., Wilkinson J.C., Mayo M.W., Duckett C.S. COMMD proteins: A novel family of structural and functional homologs of MURR1. J. Biol. Chem. 2005;280:22222–22232. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials