

BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha
- PMID: 19328063
- PMCID: PMC2818874
- DOI: 10.1016/j.molcel.2009.02.017
BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha
Abstract
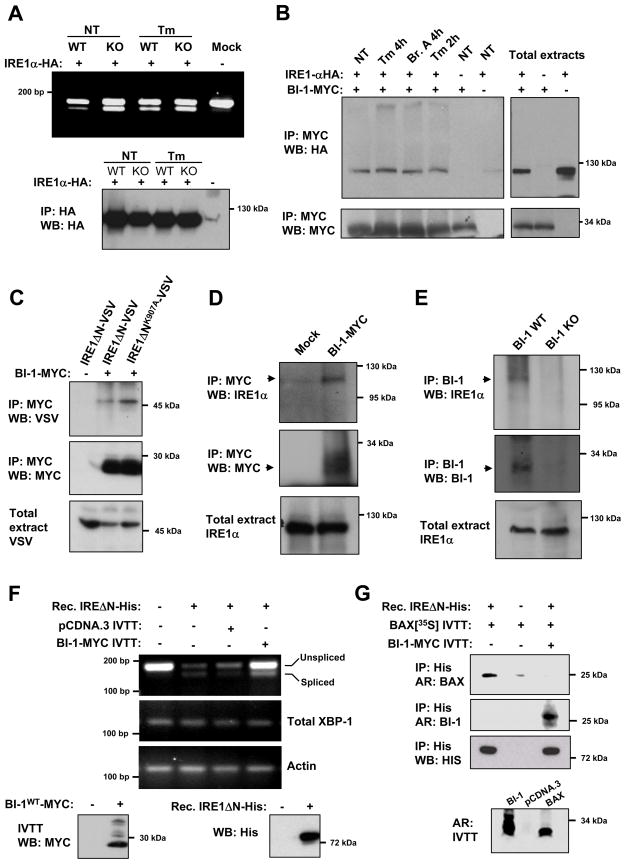
Adaptation to endoplasmic reticulum (ER) stress depends on the activation of an integrated signal transduction pathway known as the unfolded protein response (UPR). Bax inhibitor-1 (BI-1) is an evolutionarily conserved ER-resident protein that suppresses cell death. Here we have investigated the role of BI-1 in the UPR. BI-1 expression suppressed IRE1alpha activity in fly and mouse models of ER stress. BI-1-deficient cells displayed hyperactivation of the ER stress sensor IRE1alpha, leading to increased levels of its downstream target X-box-binding protein-1 (XBP-1) and upregulation of UPR target genes. This phenotype was associated with the formation of a stable protein complex between BI-1 and IRE1alpha, decreasing its ribonuclease activity. Finally, BI-1 deficiency increased the secretory activity of primary B cells, a phenomenon regulated by XBP-1. Our results suggest a role for BI-1 in early adaptive responses against ER stress that contrasts with its known downstream function in apoptosis.
Figures






Comment in
-
Intricate links between ER stress and apoptosis.Mol Cell. 2009 Mar 27;33(6):669-70. doi: 10.1016/j.molcel.2009.03.002. Mol Cell. 2009. PMID: 19328058
Similar articles
-
HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1alpha-XBP1 signaling through a physical interaction.PLoS Biol. 2010 Jul 6;8(7):e1000410. doi: 10.1371/journal.pbio.1000410. PLoS Biol. 2010. PMID: 20625543 Free PMC article.
-
Intricate links between ER stress and apoptosis.Mol Cell. 2009 Mar 27;33(6):669-70. doi: 10.1016/j.molcel.2009.03.002. Mol Cell. 2009. PMID: 19328058
-
The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis.J Clin Invest. 2005 Feb;115(2):268-81. doi: 10.1172/JCI21848. J Clin Invest. 2005. PMID: 15690081 Free PMC article.
-
The unfolded protein response: integrating stress signals through the stress sensor IRE1α.Physiol Rev. 2011 Oct;91(4):1219-43. doi: 10.1152/physrev.00001.2011. Physiol Rev. 2011. PMID: 22013210 Review.
-
Targeting the IRE1α-XBP1 branch of the unfolded protein response in human diseases.Semin Cancer Biol. 2015 Aug;33:48-56. doi: 10.1016/j.semcancer.2015.04.010. Epub 2015 May 16. Semin Cancer Biol. 2015. PMID: 25986851 Free PMC article. Review.
Cited by
-
SPHK1 deficiency protects mice from acetaminophen-induced ER stress and mitochondrial permeability transition.Cell Death Differ. 2020 Jun;27(6):1924-1937. doi: 10.1038/s41418-019-0471-x. Epub 2019 Dec 11. Cell Death Differ. 2020. PMID: 31827236 Free PMC article.
-
A crucial role for RACK1 in the regulation of glucose-stimulated IRE1alpha activation in pancreatic beta cells.Sci Signal. 2010 Jan 26;3(106):ra7. doi: 10.1126/scisignal.2000514. Sci Signal. 2010. PMID: 20103773 Free PMC article.
-
Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance.J Biol Chem. 2010 Feb 26;285(9):6198-207. doi: 10.1074/jbc.M109.056648. Epub 2009 Dec 7. J Biol Chem. 2010. PMID: 19996103 Free PMC article.
-
Deficiency in the membrane protein Tmbim3a/Grinaa initiates cold-induced ER stress and cell death by activating an intrinsic apoptotic pathway in zebrafish.J Biol Chem. 2019 Jul 26;294(30):11445-11457. doi: 10.1074/jbc.RA119.007813. Epub 2019 Jun 6. J Biol Chem. 2019. PMID: 31171717 Free PMC article.
-
Unfolded protein responses with or without unfolded proteins?Cells. 2012 Nov 1;1(4):926-50. doi: 10.3390/cells1040926. Cells. 2012. PMID: 24710536 Free PMC article.
References
-
- Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci JE, Croxton R, Krajewska M, Zapata JM, Kupiec-Weglinski JW, Farmer D, Reed JC. Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia-reperfusion injury. Proc Natl Acad Sci U S A. 2006;103:2809–2814. - PMC - PubMed
-
- Bruey JM, Bruey-Sedano N, Luciano F, Zhai D, Balpai R, Xu C, Kress CL, Bailly-Maitre B, Li X, Osterman A, Matsuzawa S, Terskikh AV, Faustin B, Reed JC. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell. 2007;129:45–56. - PubMed
-
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. - PubMed
-
- Chae HJ, Ke N, Kim HR, Chen S, Godzik A, Dickman M, Reed JC. Evolutionarily conserved cytoprotection provided by Bax Inhibitor-1 homologs from animals, plants, and yeast. Gene. 2003;323:101–113. - PubMed
-
- Chae HJ, Kim HR, Xu C, Bailly-Maitre B, Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N, Kitada S, Monosov E, Thomas M, Kress CL, Babendure JR, Tsien RY, Lipton SA, Reed JC. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell. 2004;15:355–366. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
