

Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease
- PMID: 19319544
- PMCID: PMC2853006
- DOI: 10.1007/s00401-009-0522-3
Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer's disease
Abstract
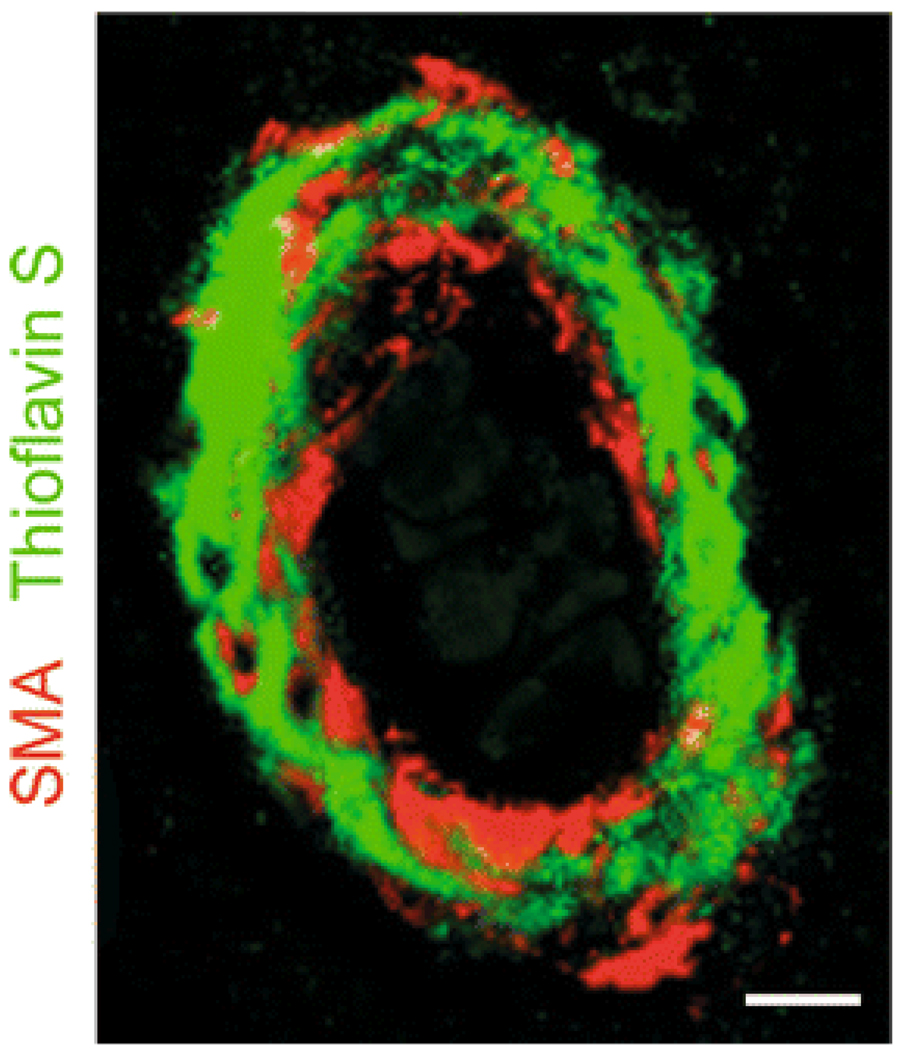
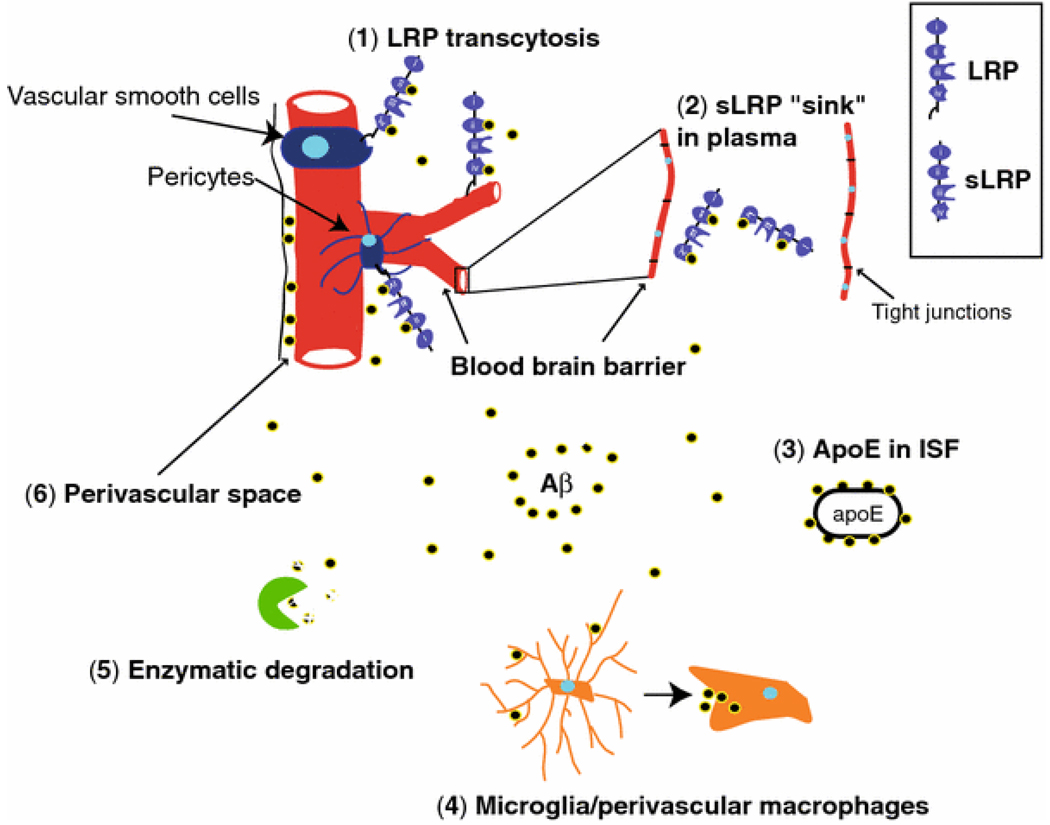
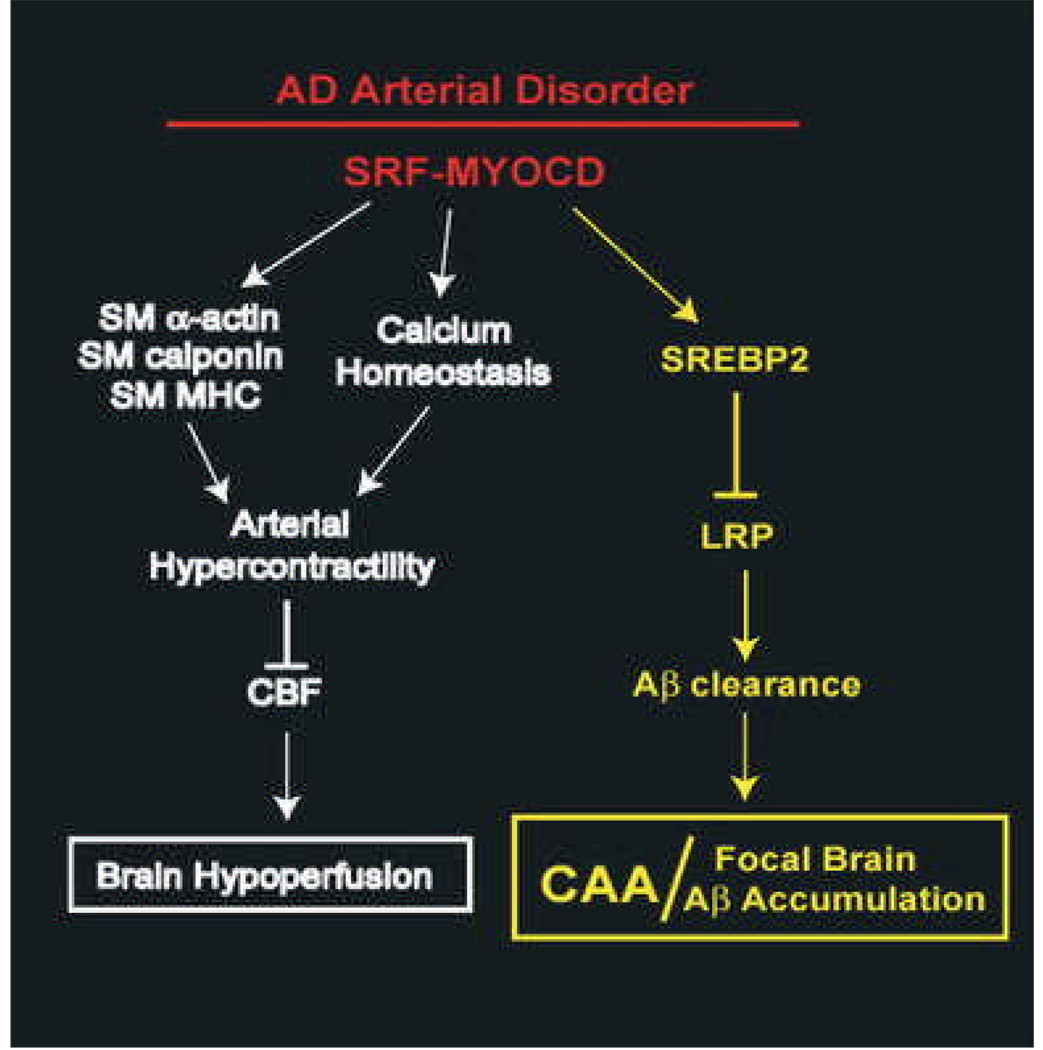
Vascular dysfunction has a critical role in Alzheimer's disease (AD). Recent data from brain imaging studies in humans and animal models suggest that cerebrovascular dysfunction may precede cognitive decline and onset of neurodegenerative changes in AD and AD models. Cerebral hypoperfusion and impaired amyloid beta-peptide (Abeta) clearance across the blood-brain barrier (BBB) may contribute to the onset and progression of dementia AD type. Decreased cerebral blood flow (CBF) negatively affects the synthesis of proteins required for memory and learning, and may eventually lead to neuritic injury and neuronal death. Impaired clearance of Abeta from the brain by the cells of the neurovascular unit may lead to its accumulation on blood vessels and in brain parenchyma. The accumulation of Abeta on the cerebral blood vessels, known as cerebral amyloid angiopathy (CAA), is associated with cognitive decline and is one of the hallmarks of AD pathology. CAA can severely disrupt the integrity of the blood vessel wall resulting in micro or macro intracerebral bleedings that exacerbates neurodegenerative process and inflammatory response and may lead to hemorrhagic stroke, respectively. Here, we review the role of the neurovascular unit and molecular mechanisms in vascular cells behind AD and CAA pathogenesis. First, we discuss apparent vascular changes, including the cerebral hypoperfusion and vascular degeneration that contribute to different stages of the disease process in AD individuals. We next discuss the role of the low-density lipoprotein receptor related protein-1 (LRP), a key Abeta clearance receptor at the BBB and along the cerebrovascular system, whose expression is suppressed early in AD. We also discuss how brain-derived apolipoprotein E isoforms may influence Abeta clearance across the BBB. We then review the role of two interacting transcription factors, myocardin and serum response factor, in cerebral vascular cells in controlling CBF responses and LRP-mediated Abeta clearance. Finally, we discuss the role of microglia and perivascular macrophages in Abeta clearance from the brain. The data reviewed here support an essential role of neurovascular and BBB mechanisms in contributing to both, onset and progression of AD.
Figures
Similar articles
-
Neurovascular dysfunction and faulty amyloid β-peptide clearance in Alzheimer disease.Cold Spring Harb Perspect Med. 2012 Oct 1;2(10):a011452. doi: 10.1101/cshperspect.a011452. Cold Spring Harb Perspect Med. 2012. PMID: 23028132 Free PMC article. Review.
-
Dysfunction of the blood-brain barrier in Alzheimer's disease: Evidence from human studies.Neuropathol Appl Neurobiol. 2022 Apr;48(3):e12782. doi: 10.1111/nan.12782. Epub 2022 Feb 2. Neuropathol Appl Neurobiol. 2022. PMID: 34823269 Review.
-
Mediators of cerebral hypoperfusion and blood-brain barrier leakiness in Alzheimer's disease, vascular dementia and mixed dementia.Brain Pathol. 2021 Jul;31(4):e12935. doi: 10.1111/bpa.12935. Epub 2021 Feb 25. Brain Pathol. 2021. PMID: 33410232 Free PMC article.
-
Endothelial Mitochondrial Dysfunction in Cerebral Amyloid Angiopathy and Alzheimer's Disease.J Alzheimers Dis. 2019;72(4):1019-1039. doi: 10.3233/JAD-190357. J Alzheimers Dis. 2019. PMID: 31306129 Free PMC article. Review.
-
Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer's disease.J Alzheimers Dis. 2013;33 Suppl 1(0 1):S87-100. doi: 10.3233/JAD-2012-129037. J Alzheimers Dis. 2013. PMID: 22751174 Free PMC article. Review.
Cited by
-
Combination of Aβ clearance and neurotrophic factors as a potential treatment for Alzheimer's disease.Neurosci Bull. 2013 Feb;29(1):111-20. doi: 10.1007/s12264-012-1287-6. Epub 2012 Nov 21. Neurosci Bull. 2013. PMID: 23179066 Free PMC article. Review.
-
Neurovascular abnormalities in brain disorders: highlights with angiogenesis and magnetic resonance imaging studies.J Biomed Sci. 2013 Jul 5;20(1):47. doi: 10.1186/1423-0127-20-47. J Biomed Sci. 2013. PMID: 23829868 Free PMC article. Review.
-
Cardiovascular risk factors, cortisol, and amyloid-β deposition in Alzheimer's Disease Neuroimaging Initiative.Alzheimers Dement. 2012 Nov;8(6):483-9. doi: 10.1016/j.jalz.2011.08.008. Alzheimers Dement. 2012. PMID: 23102118 Free PMC article.
-
Melatonin ameliorates microvessel abnormalities in the cerebral cortex and hippocampus in a rat model of Alzheimer's disease.Neural Regen Res. 2021 Apr;16(4):757-764. doi: 10.4103/1673-5374.295349. Neural Regen Res. 2021. PMID: 33063739 Free PMC article.
-
Molecular abnormalities in autopsied brain tissue from the inferior horn of the lateral ventricles of nonagenarians and Alzheimer disease patients.BMC Neurol. 2020 Aug 27;20(1):317. doi: 10.1186/s12883-020-01849-3. BMC Neurol. 2020. PMID: 32854643 Free PMC article.
References
-
- Alonzo NC, Hyman BT, Rebeck GW, Greenberg SM. Progression of cerebral amyloid angiopathy: accumulation of amyloid-beta40 in affected vessels. J Neuropathol Exp Neurol. 1998;57:353–359. doi:10.1097/00005072-199804000-00008. - PubMed
-
- Asahina M, Yoshiyama Y, Hattori T. Expression of matrix metalloproteinase-9 and urinary-type plasminogen activator in Alzheimer’s disease brain. Clin Neuropathol. 2001;20:60–63. - PubMed
-
- Attems J, Jellinger KA, Lintner F. Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol. 2005;110:222–231. doi:10.1007/s00401-005-1064-y. - PubMed
-
- Attems J, Quass M, Jellinger KA, Lintner F. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J Neurol Sci. 2007;257:49–55. doi:10.1016/j.jns.2007.01.013. - PubMed
-
- Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26:573–578. doi:10.1179/016164104225016272. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
