

Genome-wide views of chromatin structure
- PMID: 19317649
- PMCID: PMC2811691
- DOI: 10.1146/annurev.biochem.78.071107.134639
Genome-wide views of chromatin structure
Abstract
Eukaryotic genomes are packaged into a nucleoprotein complex known as chromatin, which affects most processes that occur on DNA. Along with genetic and biochemical studies of resident chromatin proteins and their modifying enzymes, mapping of chromatin structure in vivo is one of the main pillars in our understanding of how chromatin relates to cellular processes. In this review, we discuss the use of genomic technologies to characterize chromatin structure in vivo, with a focus on data from budding yeast and humans. The picture emerging from these studies is the detailed chromatin structure of a typical gene, where the typical behavior gives insight into the mechanisms and deep rules that establish chromatin structure. Important deviation from the archetype is also observed, usually as a consequence of unique regulatory mechanisms at special genomic loci. Chromatin structure shows substantial conservation from yeast to humans, but mammalian chromatin has additional layers of complexity that likely relate to the requirements of multicellularity such as the need to establish faithful gene regulatory mechanisms for cell differentiation.
Figures
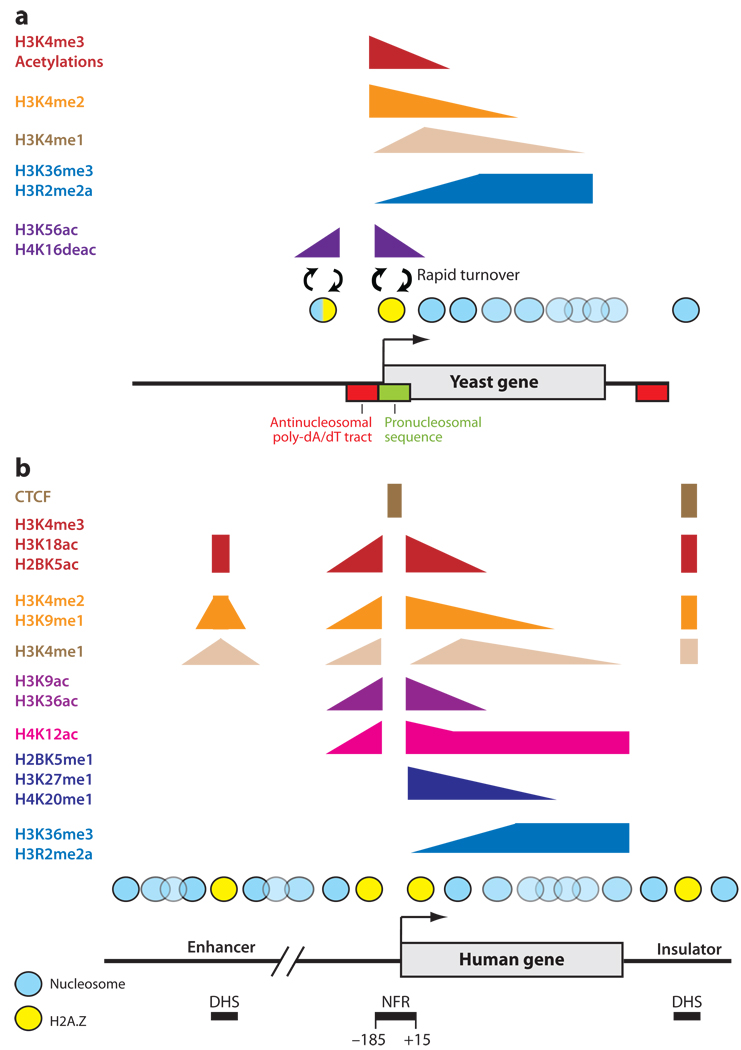
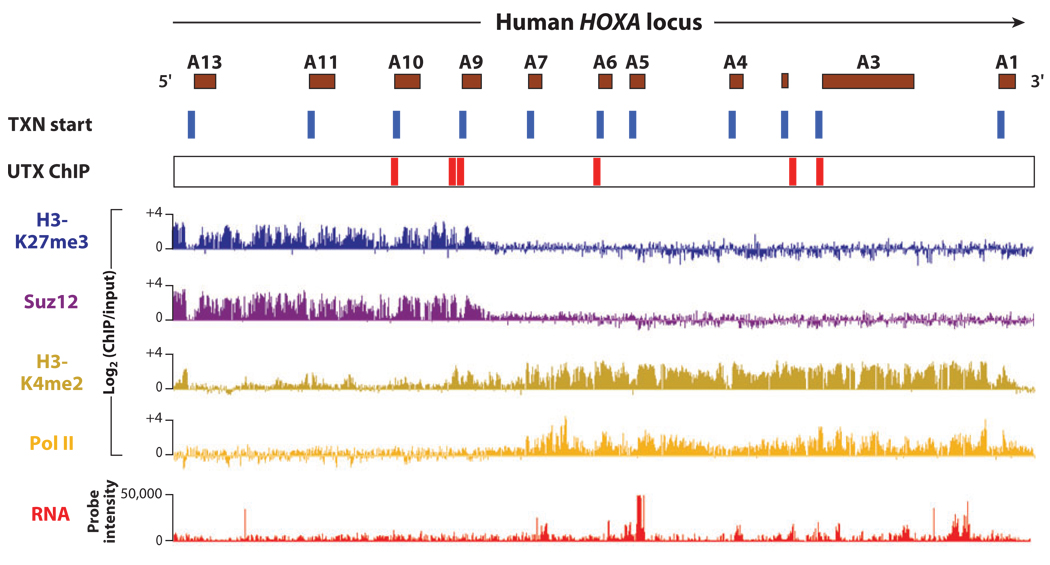
Similar articles
-
Genome-wide in vitro reconstitution of yeast chromatin with in vivo-like nucleosome positioning.Methods Enzymol. 2012;513:205-32. doi: 10.1016/B978-0-12-391938-0.00009-4. Methods Enzymol. 2012. PMID: 22929771
-
Chromatin modifications and chromatin remodeling during DNA repair in budding yeast.Curr Opin Genet Dev. 2013 Apr;23(2):166-73. doi: 10.1016/j.gde.2012.11.015. Epub 2013 Apr 17. Curr Opin Genet Dev. 2013. PMID: 23602331 Review.
-
Quantitative genetic analysis in Saccharomyces cerevisiae using epistatic miniarray profiles (E-MAPs) and its application to chromatin functions.Methods. 2006 Dec;40(4):344-52. doi: 10.1016/j.ymeth.2006.07.034. Methods. 2006. PMID: 17101447
-
Chromatin and transcription in Saccharomyces cerevisiae.FEMS Microbiol Rev. 1999 Jul;23(4):503-23. doi: 10.1111/j.1574-6976.1999.tb00410.x. FEMS Microbiol Rev. 1999. PMID: 10422263 Review.
-
Opening windows to the genome.Cell. 2009 May 1;137(3):400-2. doi: 10.1016/j.cell.2009.04.026. Cell. 2009. PMID: 19410536
Cited by
-
Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements.Genome Res. 2012 Sep;22(9):1735-47. doi: 10.1101/gr.136366.111. Genome Res. 2012. PMID: 22955985 Free PMC article.
-
Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells.Cell. 2011 Dec 23;147(7):1498-510. doi: 10.1016/j.cell.2011.11.054. Cell. 2011. PMID: 22196727 Free PMC article.
-
DNA looping-dependent targeting of a chromatin remodeling factor.Cell Cycle. 2013 Jun 15;12(12):1809-10. doi: 10.4161/cc.25114. Epub 2013 May 24. Cell Cycle. 2013. PMID: 23708514 Free PMC article. No abstract available.
-
Linking genome to epigenome.Wiley Interdiscip Rev Syst Biol Med. 2012 May-Jun;4(3):297-309. doi: 10.1002/wsbm.1165. Epub 2012 Feb 17. Wiley Interdiscip Rev Syst Biol Med. 2012. PMID: 22344857 Free PMC article. Review.
-
Target gene context influences the transcriptional requirement for the KAT3 family of CBP and p300 histone acetyltransferases.Epigenetics. 2010 Jan 1;5(1):9-15. doi: 10.4161/epi.5.1.10449. Epub 2010 Jan 27. Epigenetics. 2010. PMID: 20110770 Free PMC article. Review.
References
-
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. - PubMed
-
- Olins AL, Olins DE. Spheroid chromatin units (v bodies) Science. 1974;183:330–332. - PubMed
-
- Ptashne M. On the use of the word ‘epigenetic’. Curr. Biol. 2007;17:R233–R236. - PubMed
-
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods. 2006;3:511–518. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
