

Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons
- PMID: 19288225
- PMCID: PMC2677622
- DOI: 10.1007/s12017-009-8058-1
Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons
Abstract
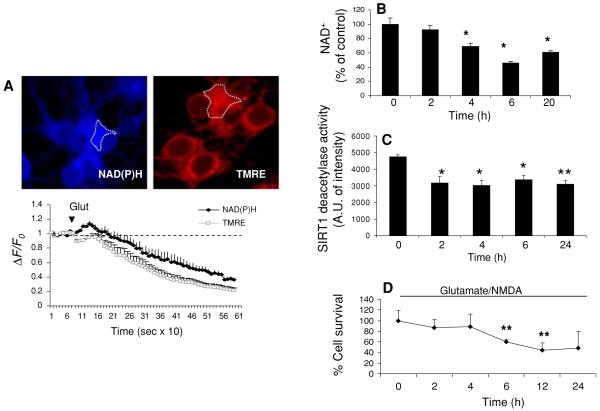
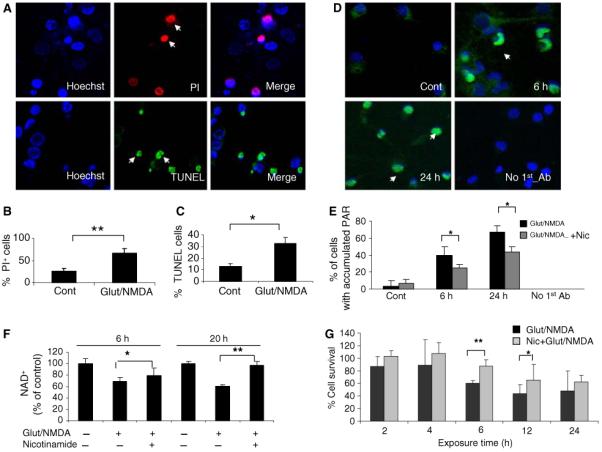
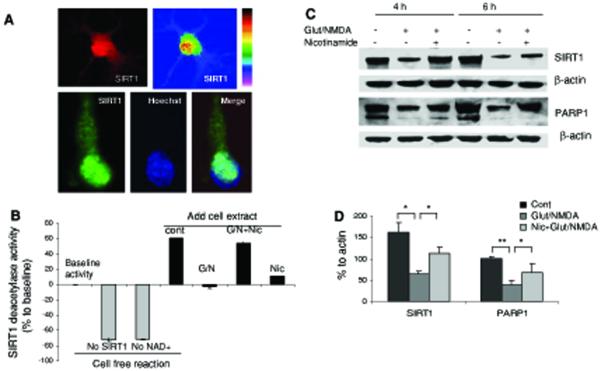
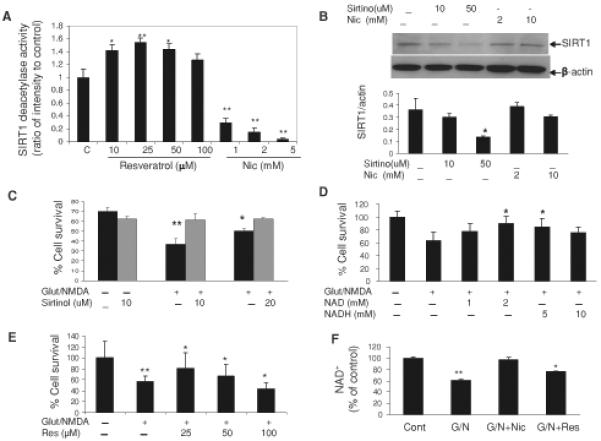
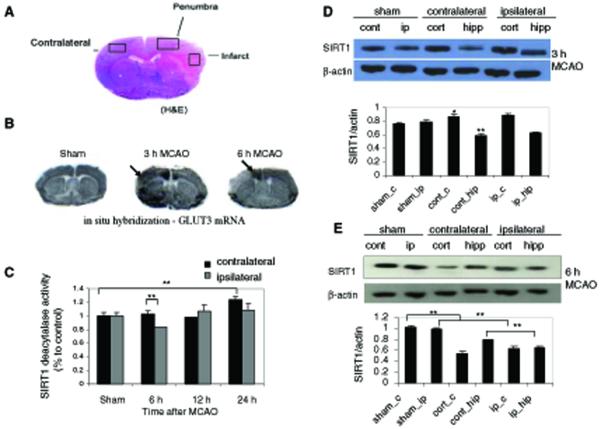
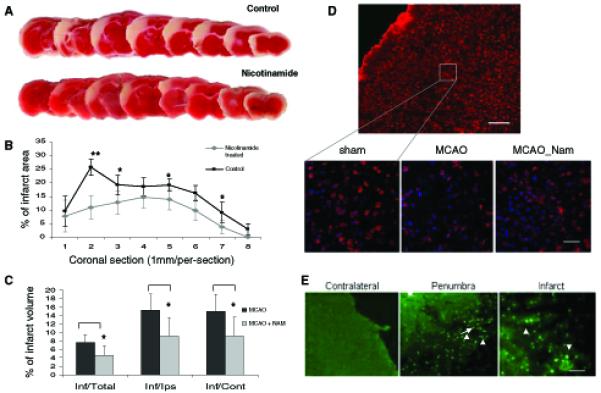
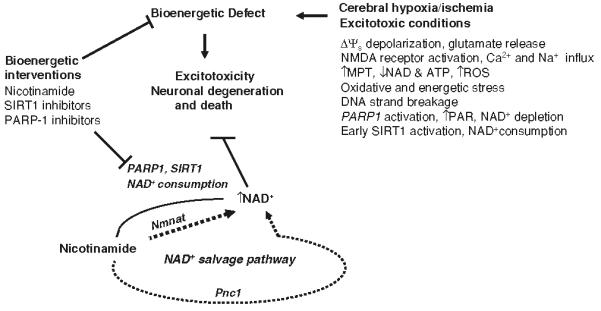
Neurons require large amounts of energy to support their survival and function, and are therefore susceptible to excitotoxicity, a form of cell death involving bioenergetic stress that may occur in several neurological disorders including stroke and Alzheimer's disease. Here we studied the roles of NAD(+) bioenergetic state, and the NAD(+)-dependent enzymes SIRT1 and PARP-1, in excitotoxic neuronal death in cultured neurons and in a mouse model of focal ischemic stroke. Excitotoxic activation of NMDA receptors induced a rapid decrease of cellular NAD(P)H levels and mitochondrial membrane potential. Decreased NAD(+) levels and poly (ADP-ribose) polymer (PAR) accumulation in nuclei were relatively early events (<4 h) that preceded the appearance of propidium iodide- and TUNEL-positive cells (markers of necrotic cell death and DNA strand breakage, respectively) which became evident by 6 h. Nicotinamide, an NAD(+) precursor and an inhibitor of SIRT1 and PARP1, inhibited SIRT1 deacetylase activity without affecting SIRT1 protein levels. NAD(+) levels were preserved and PAR accumulation and neuronal death induced by excitotoxic insults were attenuated in nicotinamide-treated cells. Treatment of neurons with the SIRT1 activator resveratrol did not protect them from glutamate/NMDA-induced NAD(+) depletion and death. In a mouse model of focal cerebral ischemic stroke, NAD(+) levels were decreased in both the contralateral and ipsilateral cortex 6 h after the onset of ischemia. Stroke resulted in dynamic changes of SIRT1 protein and activity levels which varied among brain regions. Administration of nicotinamide (200 mg/kg, i.p.) up to 1 h after the onset of ischemia elevated brain NAD(+) levels and reduced ischemic infarct size. Our findings demonstrate that the NAD(+) bioenergetic state is critical in determining whether neurons live or die in excitotoxic and ischemic conditions, and suggest a potential therapeutic benefit in stroke of agents that preserve cellular NAD(+) levels. Our data further suggest that, SIRT1 is linked to bioenergetic state and stress responses in neurons, and that under conditions of reduced cellular energy levels SIRT1 enzyme activity may consume sufficient NAD(+) to nullify any cell survival-promoting effects of its deacetylase action on protein substrates.
Figures
Similar articles
-
Preventing NAD(+) depletion protects neurons against excitotoxicity: bioenergetic effects of mild mitochondrial uncoupling and caloric restriction.Ann N Y Acad Sci. 2008 Dec;1147:275-82. doi: 10.1196/annals.1427.028. Ann N Y Acad Sci. 2008. PMID: 19076449 Free PMC article.
-
Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway.Neuroscience. 2009 Mar 31;159(3):993-1002. doi: 10.1016/j.neuroscience.2009.01.017. Epub 2009 Jan 19. Neuroscience. 2009. PMID: 19356683 Free PMC article.
-
Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture.PLoS One. 2011 Mar 1;6(3):e14731. doi: 10.1371/journal.pone.0014731. PLoS One. 2011. PMID: 21390294 Free PMC article.
-
Resveratrol and ischemic preconditioning in the brain.Curr Med Chem. 2008;15(15):1545-51. doi: 10.2174/092986708784638861. Curr Med Chem. 2008. PMID: 18537630 Review.
-
NAD+ metabolism and NAD(+)-dependent enzymes: promising therapeutic targets for neurological diseases.Curr Drug Targets. 2012 Feb;13(2):222-9. doi: 10.2174/138945012799201711. Curr Drug Targets. 2012. PMID: 22204321 Review.
Cited by
-
Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease.J Clin Invest. 2013 Jul;123(7):3084-98. doi: 10.1172/JCI64401. Epub 2013 Jun 17. J Clin Invest. 2013. PMID: 23778143 Free PMC article.
-
Nicotinamide, NAD(P)(H), and Methyl-Group Homeostasis Evolved and Became a Determinant of Ageing Diseases: Hypotheses and Lessons from Pellagra.Curr Gerontol Geriatr Res. 2012;2012:302875. doi: 10.1155/2012/302875. Epub 2012 Mar 21. Curr Gerontol Geriatr Res. 2012. PMID: 22536229 Free PMC article.
-
The role of the tryptophan-NAD + pathway in a mouse model of severe malnutrition induced liver dysfunction.Nat Commun. 2022 Dec 8;13(1):7576. doi: 10.1038/s41467-022-35317-y. Nat Commun. 2022. PMID: 36481684 Free PMC article.
-
Genetic control of necrosis - another type of programmed cell death.Curr Opin Cell Biol. 2010 Dec;22(6):882-8. doi: 10.1016/j.ceb.2010.09.002. Curr Opin Cell Biol. 2010. PMID: 20889324 Free PMC article. Review.
-
Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders.Antioxid Redox Signal. 2010 Dec 1;13(11):1763-811. doi: 10.1089/ars.2009.3074. Epub 2010 Aug 28. Antioxid Redox Signal. 2010. PMID: 20446769 Free PMC article. Review.
References
-
- Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circulation Research. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. - PubMed
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, et al. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi:10.1016/ 0896-6273(95)90186-8. - PubMed
-
- Antzoulatos EG, Byrne JH. Learning insights transmitted by glutamate. Trends in Neurosciences. 2004;27:555–560. doi:10.1016/j.tins.2004.06.009. - PubMed
-
- Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD+biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–1013. doi:10.1126/science.1098014. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
