

Transcription factor binding sites are genetic determinants of retroviral integration in the human genome
- PMID: 19238208
- PMCID: PMC2642719
- DOI: 10.1371/journal.pone.0004571
Transcription factor binding sites are genetic determinants of retroviral integration in the human genome
Abstract
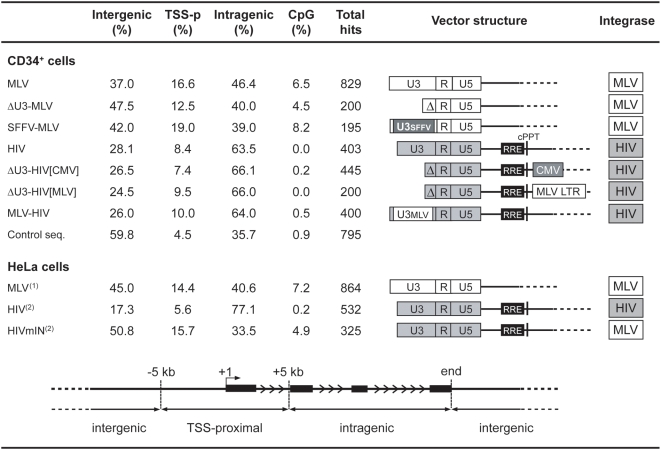
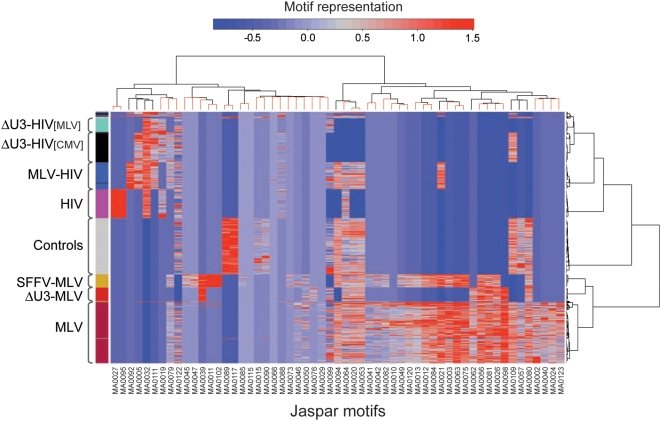
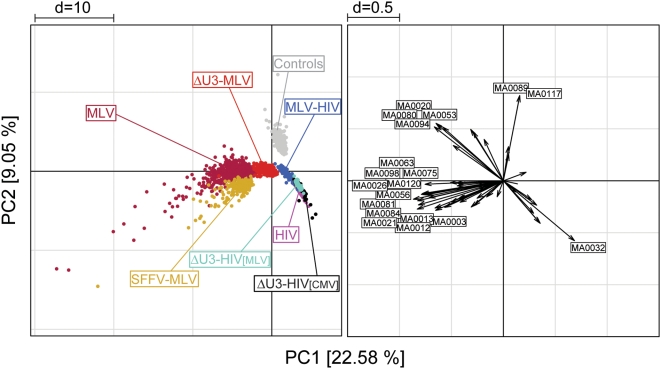
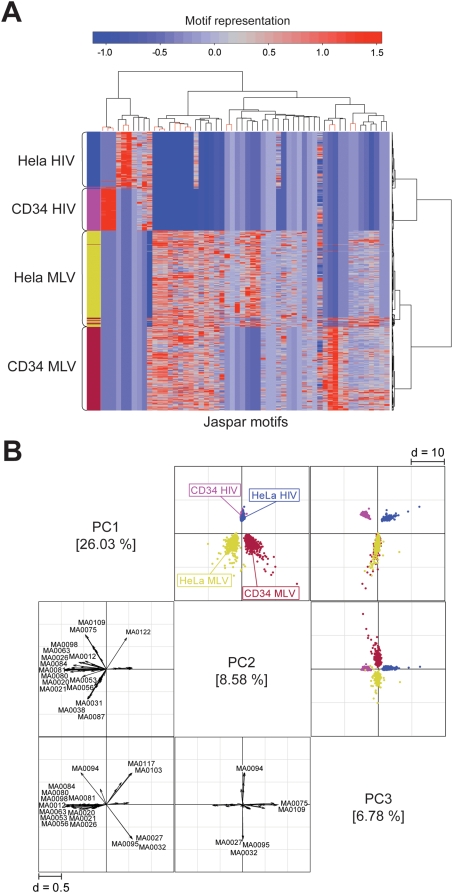
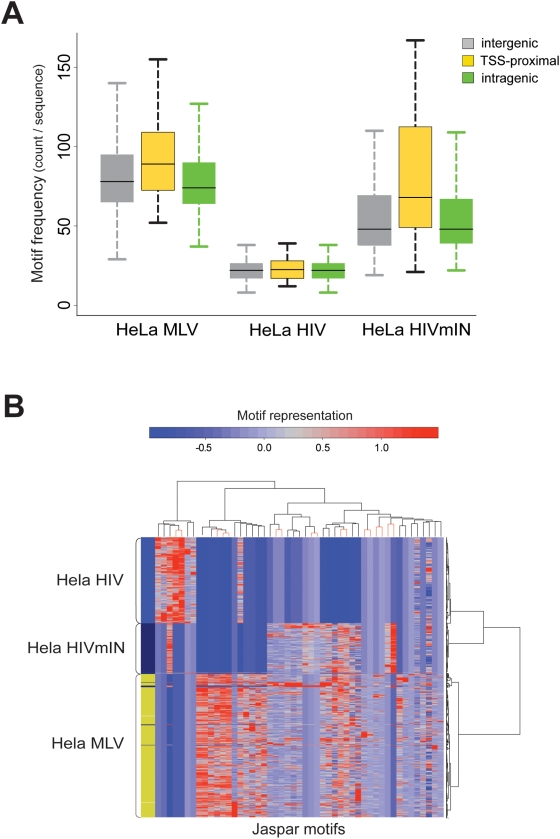
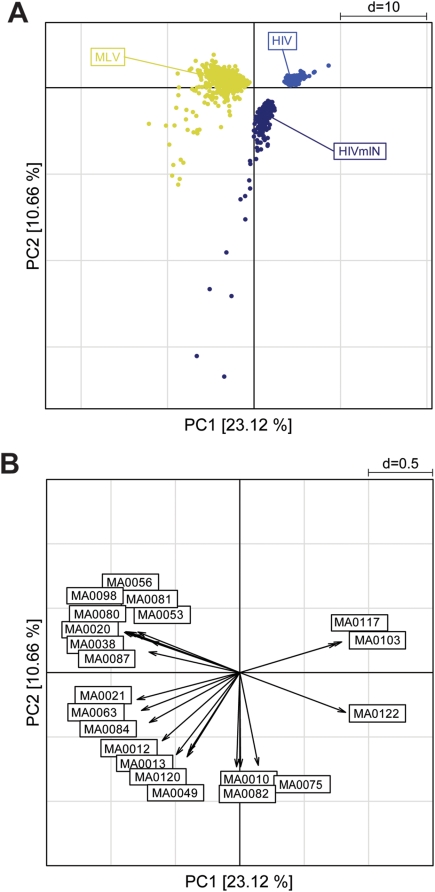
Gamma-retroviruses and lentiviruses integrate non-randomly in mammalian genomes, with specific preferences for active chromatin, promoters and regulatory regions. Gene transfer vectors derived from gamma-retroviruses target at high frequency genes involved in the control of growth, development and differentiation of the target cell, and may induce insertional tumors or pre-neoplastic clonal expansions in patients treated by gene therapy. The gene expression program of the target cell is apparently instrumental in directing gamma-retroviral integration, although the molecular basis of this phenomenon is poorly understood. We report a bioinformatic analysis of the distribution of transcription factor binding sites (TFBSs) flanking >4,000 integrated proviruses in human hematopoietic and non-hematopoietic cells. We show that gamma-retroviral, but not lentiviral vectors, integrate in genomic regions enriched in cell-type specific subsets of TFBSs, independently from their relative position with respect to genes and transcription start sites. Analysis of sequences flanking the integration sites of Moloney leukemia virus (MLV)- and human immunodeficiency virus (HIV)-derived vectors carrying mutations in their long terminal repeats (LTRs), and of HIV vectors packaged with an MLV integrase, indicates that the MLV integrase and LTR enhancer are the viral determinants of the selection of TFBS-rich regions in the genome. This study identifies TFBSs as differential genomic determinants of retroviral target site selection in the human genome, and suggests that transcription factors binding the LTR enhancer may synergize with the integrase in tethering retroviral pre-integration complexes to transcriptionally active regulatory regions. Our data indicate that gamma-retroviruses and lentiviruses have evolved dramatically different strategies to interact with the host cell chromatin, and predict a higher risk in using gamma-retroviral vs. lentiviral vectors for human gene therapy applications.
Conflict of interest statement
Figures


Similar articles
-
Retroviral DNA integration: viral and cellular determinants of target-site selection.PLoS Pathog. 2006 Jun;2(6):e60. doi: 10.1371/journal.ppat.0020060. Epub 2006 Jun 23. PLoS Pathog. 2006. PMID: 16789841 Free PMC article.
-
Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses.J Virol. 2007 Jun;81(12):6731-41. doi: 10.1128/JVI.02752-06. Epub 2007 Apr 4. J Virol. 2007. PMID: 17409138 Free PMC article.
-
Deletion of the LTR enhancer/promoter has no impact on the integration profile of MLV vectors in human hematopoietic progenitors.PLoS One. 2013;8(1):e55721. doi: 10.1371/journal.pone.0055721. Epub 2013 Jan 31. PLoS One. 2013. PMID: 23383272 Free PMC article.
-
Integration site selection by retroviral vectors: molecular mechanism and clinical consequences.Hum Gene Ther. 2008 Jun;19(6):557-68. doi: 10.1089/hum.2007.148. Hum Gene Ther. 2008. PMID: 18533894 Free PMC article. Review.
-
Determinants of Retroviral Integration and Implications for Gene Therapeutic MLV-Based Vectors and for a Cure for HIV-1 Infection.Viruses. 2022 Dec 21;15(1):32. doi: 10.3390/v15010032. Viruses. 2022. PMID: 36680071 Free PMC article. Review.
Cited by
-
Early events of HIV-1 infection: can signaling be the next therapeutic target?J Neuroimmune Pharmacol. 2011 Jun;6(2):269-83. doi: 10.1007/s11481-011-9268-5. Epub 2011 Mar 5. J Neuroimmune Pharmacol. 2011. PMID: 21373988 Review.
-
The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites.Cell Rep. 2013 Nov 27;5(4):886-94. doi: 10.1016/j.celrep.2013.09.040. Epub 2013 Oct 31. Cell Rep. 2013. PMID: 24183673 Free PMC article.
-
Integrated analysis of microRNA and transcription factors in the bone marrow of patients with acute monocytic leukemia.Oncol Lett. 2021 Jan;21(1):50. doi: 10.3892/ol.2020.12311. Epub 2020 Nov 18. Oncol Lett. 2021. PMID: 33281961 Free PMC article.
-
Insight in HIV Integration Site Selection Provides a Block-and-Lock Strategy for a Functional Cure of HIV Infection.Viruses. 2018 Dec 26;11(1):12. doi: 10.3390/v11010012. Viruses. 2018. PMID: 30587760 Free PMC article. Review.
-
The next generation of transcription factor binding site prediction.PLoS Comput Biol. 2013;9(9):e1003214. doi: 10.1371/journal.pcbi.1003214. Epub 2013 Sep 5. PLoS Comput Biol. 2013. PMID: 24039567 Free PMC article.
References
-
- Coffin JM, Huges SH, Varmus HE. Retroviruses. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. - PubMed
-
- Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, et al. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. - PubMed
-
- Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. - PubMed
-
- Bushman FD. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell. 2003;115:135–138. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
