

Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis
- PMID: 19135240
- PMCID: PMC2638021
- DOI: 10.1016/j.cell.2008.11.018
Regulation of PKD by the MAPK p38delta in insulin secretion and glucose homeostasis
Abstract
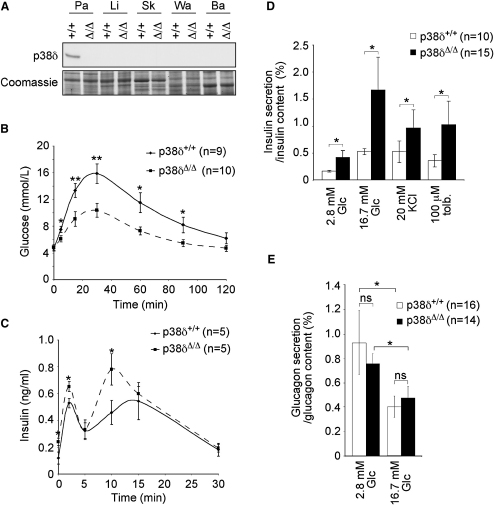
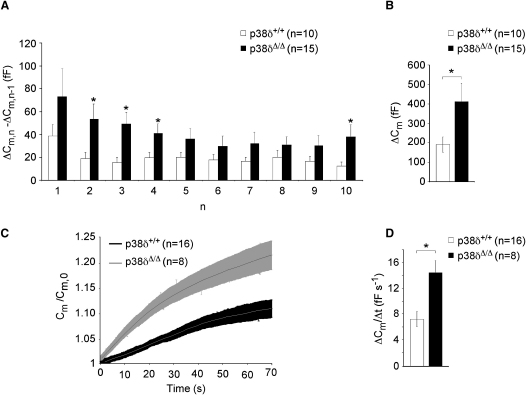
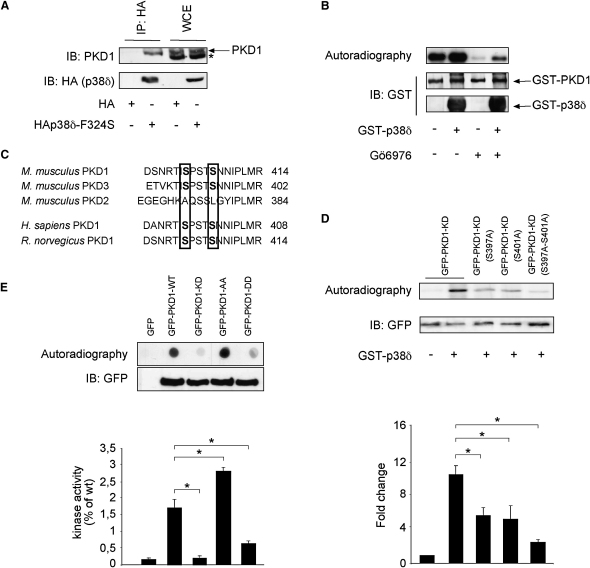
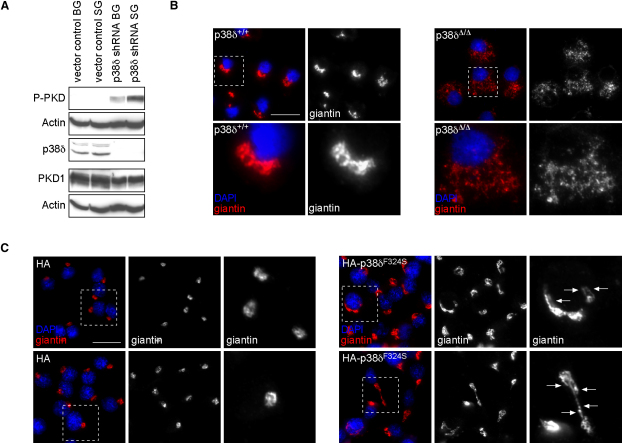
Dysfunction and loss of insulin-producing pancreatic beta cells represent hallmarks of diabetes mellitus. Here, we show that mice lacking the mitogen-activated protein kinase (MAPK) p38delta display improved glucose tolerance due to enhanced insulin secretion from pancreatic beta cells. Deletion of p38delta results in pronounced activation of protein kinase D (PKD), the latter of which we have identified as a pivotal regulator of stimulated insulin exocytosis. p38delta catalyzes an inhibitory phosphorylation of PKD1, thereby attenuating stimulated insulin secretion. In addition, p38delta null mice are protected against high-fat-feeding-induced insulin resistance and oxidative stress-mediated beta cell failure. Inhibition of PKD1 reverses enhanced insulin secretion from p38delta-deficient islets and glucose tolerance in p38delta null mice as well as their susceptibility to oxidative stress. In conclusion, the p38delta-PKD pathway integrates regulation of the insulin secretory capacity and survival of pancreatic beta cells, pointing to a pivotal role for this pathway in the development of overt diabetes mellitus.
Figures
Comment in
-
p38delta and PKD1: kinase switches for insulin secretion.Cell. 2009 Jan 23;136(2):209-10. doi: 10.1016/j.cell.2009.01.005. Cell. 2009. PMID: 19167323
Similar articles
-
Impaired insulin exocytosis in chronic hepatitis C infection: contributory role of p38δ MAPK-protein kinase D-golgi complex axis.Clin Sci (Lond). 2020 Jun 26;134(12):1449-1456. doi: 10.1042/CS20200686. Clin Sci (Lond). 2020. PMID: 32556178
-
Chronic hepatitis C virus infection impairs insulin secretion by regulation of p38δ MAPK-dependent exocytosis in pancreatic β-cells.Clin Sci (Lond). 2020 Mar 13;134(5):529-542. doi: 10.1042/CS20190900. Clin Sci (Lond). 2020. PMID: 32100852
-
p38delta and PKD1: kinase switches for insulin secretion.Cell. 2009 Jan 23;136(2):209-10. doi: 10.1016/j.cell.2009.01.005. Cell. 2009. PMID: 19167323
-
Signal transduction in pancreatic beta-cells: regulation of insulin secretion by information flow in the phospholipase C/protein kinase C pathway.Front Biosci. 1997 Mar 15;2:d160-72. doi: 10.2741/a180. Front Biosci. 1997. PMID: 9159224 Review.
-
Diacylglycerol Signaling Pathway in Pancreatic β-Cells: An Essential Role of Diacylglycerol Kinase in the Regulation of Insulin Secretion.Biol Pharm Bull. 2015;38(5):669-73. doi: 10.1248/bpb.b15-00060. Biol Pharm Bull. 2015. PMID: 25947912 Review.
Cited by
-
A first-in-kind MAPK13 inhibitor that can correct stem cell reprogramming and post-injury disease.bioRxiv [Preprint]. 2024 Aug 22:2024.08.21.608990. doi: 10.1101/2024.08.21.608990. bioRxiv. 2024. PMID: 39229202 Free PMC article. Preprint.
-
PPARβ/δ affects pancreatic β cell mass and insulin secretion in mice.J Clin Invest. 2012 Nov;122(11):4105-17. doi: 10.1172/JCI42127. Epub 2012 Oct 24. J Clin Invest. 2012. PMID: 23093780 Free PMC article.
-
RNAi screening reveals a large signaling network controlling the Golgi apparatus in human cells.Mol Syst Biol. 2012;8:629. doi: 10.1038/msb.2012.59. Mol Syst Biol. 2012. PMID: 23212246 Free PMC article.
-
Atypical Antipsychotics and Metabolic Syndrome: From Molecular Mechanisms to Clinical Differences.Pharmaceuticals (Basel). 2021 Mar 8;14(3):238. doi: 10.3390/ph14030238. Pharmaceuticals (Basel). 2021. PMID: 33800403 Free PMC article. Review.
-
p38γ and p38δ Mitogen Activated Protein Kinases (MAPKs), New Stars in the MAPK Galaxy.Front Cell Dev Biol. 2016 Apr 14;4:31. doi: 10.3389/fcell.2016.00031. eCollection 2016. Front Cell Dev Biol. 2016. PMID: 27148533 Free PMC article. Review.
References
-
- Adams R.H., Porras A., Alonso G., Jones M., Vintersten K., Panelli S., Valladares A., Perez L., Klein R., Nebreda A.R. Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell. 2000;6:109–116. - PubMed
-
- Ammala C., Eliasson L., Bokvist K., Berggren P.O., Honkanen R.E., Sjoholm A., Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 1994;91:4343–4347. - PMC - PubMed
-
- Askari N., Diskin R., Avitzour M., Capone R., Livnah O., Engelberg D. Hyperactive variants of p38alpha induce, whereas hyperactive variants of p38gamma suppress, activating protein 1-mediated transcription. J. Biol. Chem. 2007;282:91–99. - PubMed
-
- Aston-Mourney K., Proietto J., Morahan G., Andrikopoulos S. Too much of a good thing: why it is bad to stimulate the beta cell to secrete insulin. Diabetologia. 2008;51:540–545. - PubMed
-
- Bard F., Malhotra V. The formation of TGN-to-plasma-membrane transport carriers. Annu. Rev. Cell Dev. Biol. 2006;22:439–455. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
